Электролитическая диссоциация — процесс распада электролита на ионы при его растворении или плавлении.
Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Аррениус придерживался физической теории растворов, не учитывал взаимодействие электролита с водой и считал, что в растворах находятся свободные ионы. Русские химики И. А. Каблукови В. А. Кистяковский применили для объяснения электролитической диссоциации химическую теорию растворов Д. И. Менделеева и доказали, что при растворении электролита происходит его химическое взаимодействие с водой, в результате которого электролит диссоциирует на ионы.
Классическая теория электролитической диссоциации основана на предположении о неполной диссоциации растворённого вещества, характеризуемой степенью диссоциации α, т. е. долей распавшихся молекул электролита. Динамическое равновесие между недиссоциированными молекулами и ионами описывается законом действующих масс .
Вещества, распадающиеся на ионы, называют электролитами. Электролиты – вещества с ионной или сильно ковалентной связью: кислоты, основания, соли. остальные вещества – неэлектролиты; к ним относятся вещества с неполярной или слабо полярной ковалентной связью; например, многие органические соединения.
Основные положения ТЭД (Теории электролитической диссоциации):
Молекулы распадаются на положительно и отрицательно заряженные ионы (простые и сложные).
Под действием электрического тока катионы (положительно заряженные ионы движутся к катоду(-), а анионы (отрицательно заряженные ионы) к аноду(+)
Степень диссоциации зависит от природы вещества и растворителя, концентрации, температуры.
Если степень диссоциации зависит от природы вещества, то можно судить, что существует разграничение между определёнными группами веществ.
Большая степень диссоциации присуща сильным электролитам (большинству оснований, солям, многим кислотам). Стоит учесть, что распад на ионы – обратимая реакция. Так же стоит сказать, что в данной теме не будут разобраны примеры диссоциации двойных и основных солей, их диссоциация описана в теме “соли”.
Примеры сильных электролитов:
NaOH, K2SO4, HClO4
Уравнения диссоциации:
NaOH⇄Na + +OH —
Количественной характеристикой силы электролитов является степень диссоциации (α) – отношение молярной концентрации продиссоциировавшего электролита к его общей молярной концентрации в растворе.
Степень диссоциации выражается в долях единицы или в процентах. Интервал значений – от 0 до 100%.
α = 0% относится к неэлектролитам (диссоциация отсутствует)
У каждой ступени диссоциации своя степень диссоциации.
Например, диссоциация солей CuCl2, HgCl2:
CuCl2⇄Cu 2+ +2Cl — диссоциация протекает полностью
А в случае с хлоридом ртути диссоциация идёт неполностью и то не до конца.
Возвращаясь же к раствору серной кислоты, стоит сказать, что степень диссоциации обеих ступеней разбавленной кислоты гораздо больше, чем у концентрированной. При диссоциации концентрированного раствора очень много молекул вещества и большая концентрация гидроанионов HSO4 — .
У многоосновных кислот и многокислотных оснований диссоциация идёт в несколько ступеней (в зависимости от основности).
Перечислим сильные и слабые кислоты и приступим к уравнениям ионного обмена:
Сильные кислоты ( HCl, HBr, HI, HClO3, HBrO3, HIO3, HClO4, H2SO4, H2SeO4,HNO3, HMnO4, H2Cr2O7)
Химические реакции в растворах и расплавах электролитов протекают с участием ионов. В таких реакциях степени окисления элементов не изменяются, и сами реакции называются реакциями ионного обмена.
Реакции ионного обмена будут протекать до конца (необратимо) , если образуются малорастворимые или практически нерастворимые вещества (они выпадают в осадок), летучие вещества (выделяются в виде газов) или слабые электролиты (например, вода).
Реакции ионного обмена принято писать в три стадии:
1. Молекулярное уравнение
2. Полное ионное уравнение
3. Сокращенное ионное уравнение
При написании обязательно указывать осадки и газы, а так же руководствоваться таблицей растворимости.
Реакции, где все реагенты и продукты получились растворимые в воде, не протекают.
Сокращённое ионное уравнение получается с помощью вычёркивания одинаковых ионов из обеих частей полного ионного уравнения.
Если реакция ионного обмена идёт между двумя солями с образованием осадка, то следует брать два хорошо растворимых реагента. То есть, реакция ионного обмена пойдёт если растворимость реагентов будет выше, чем у одного из продуктов.
Иногда при написании реакций ионного обмена пропускают полное ионное уравнение и сразу пишут сокращенное.
Для получения осадка малорастворимого вещества всегда надо выбирать хорошо растворимые реагенты в их концентрированных растворах.
Например:
2KF+FeCl2→FeF2↓+2KCl
Данные правила подбора реагентов для осаждения продуктов справедливы только для солей.
студентам и школьникам
Шпаргалки по электрохимии — Теория электролитической диссоциации Аррениуса
Cмотрите так же.
Теория электролитической диссоциации Аррениуса. Закон разбавления Оствальда. Степень диссоциации, константа диссоциации. Недостатки теории Аррениуса.
Теория электролитической диссоциации Аррениуса
Для электролитов коллигативные свойства растворов (понижение температуры замерзания, повышение температуры кипения, понижение давления пара растворителя над раствором и осмотическое давление) значительно больше соответствующих величин для неэлектролитов. В уравнение для осмотического давления p Вант-Гофф ввел эмпирический коэффициент i > 1, физический смысл которого стал понятен с появлением теории электролитической диссоциации:
Теория электролитической диссоциации была предложена Аррениусом (1884-1887), развившим отдельные высказывания ряда ученых.
Основные положения теории Аррениуса :
1. Соли, кислоты, основания при растворении в воде и некоторых других полярных растворителях частично или полностью распадаются (диссоциируют) на ионы . Эти ионы существуют в растворе независимо от того, проходит через раствор электрический ток или нет. Вследствие этого число независимо движущихся частиц растворенного вещества больше, чем при отсутствии диссоциации, а величины коллигативных свойств растворов возрастают прямо пропорционально числу частиц. Ионы представляют собой заряженные частицы, которые состоят или из отдельных атомов, или из группы атомов. Предполагается, что ионы в растворе ведут себя подобно молекулам идеального газа, то есть не взаимодействуют друг с другом.
2. Наряду с процессом диссоциации в растворе идет обратный процесс — ассоциация ионов в молекулы. Таким образом, диссоциация молекул на ионы является неполной , поэтому в качестве меры электролитической диссоциации Аррениус ввел величину степени диссоциации a , определяемую как долю молекул, распавшихся на ионы:
Для любой обратимой реакции электролитической диссоциации
сумма n + + n – равна общему числу n ионов, образующихся при диссоциации одной молекулы; связь с коэффициентом Вант-Гоффа i дается уравнением
i = 1 + ( n + + n – — 1) × a = 1 + ( n — 1) × a .
Определив коэффициент i , можно по этому уравнению вычислить степень диссоциации a , если известна величина n .
Коэффициент i показывает, во сколько раз увеличивается общая молярная конценрация частиц в растворе за счет диссоциации электролита. По мере увеличения разведения коэффициент Вант-Гоффа приближается к простому целому числу (2, 3, 4 — в зависимости от числа ионов, образующихся из одной молекулы вещества).
3. Диссоциация растворенных веществ на ионы подчиняется тем же законам химического равновесия, что и другие реакции , в частности, закону действующих масс
где Кд,с — константа диссоциации, выраженная через концентрации, или так называемая классическая константа диссоциации.
Диссоциация сильных электролитов равна 100% или почти 100%, так что концентрации ионов можно считать равными молярности растворенного вещества, умноженной на n + ( n – ):
При диссоциации слабого электролита устанавливается равновесие между недиссоциированными молекулами и ионами. Рассмотрим простейший пример , когда молекула распадается только на два иона:
с — a с a с a с (равновесные концентрации)
Последнее равенство является простейшей формой закона разведения Оствальда (1888), поскольку величина V = 1/ с , л/моль, называется разведением.
Чем больше Кд,с , тем выше степень диссоциации. Таким образом, величина Кд,с может служить мерой силы кислоты, то есть мерой кислотности. Для электролитов средней силы (Н3РО4 — первая ступень, Са(ОН)2, СНС l 2СООН) значения Кд,с лежат в пределах от 10 –2 до 10 –4 ; для слабых электролитов (СН3СООН, N Н4ОН) Кд,с = 10 –5 — 10 –9 ; при Кд,с 10 –10 электролит считается очень слабым (Н2О, С6Н5ОН, С6Н5 N Н2, НС N ).
Зная константу диссоциации, можно рассчитать степень диссоциации в зависимости от концентрации электролита. Решая квадратное уравнение и учитывая, что a > 0, получим
Как следует из данного уравнения, при условии Кд,с > > 4 с , a ® 1, то есть электролит становится полностью диссоциированным. С другой стороны, при малых Кд,с (обычно 10 –5 ) и при не очень низких конценрациях, когда Кд,с 4 с, величиной a можно пренебречь по сравнению с 1 в знаменателе закона разведения Оствальда, и формулы примут вид
Кд,с = a 2 с ; a = .
Вышеприведенные соотношения применимы только для растворов симметричных бинарных электролитов (то есть если одна молекула электролита дает один катион и один анион). Если электролит распадается больше чем на два иона, то зависимость Кд,с от a усложняется:
Са Cl 2 Û Ca 2+ + 2 Cl –
с (1- a ) a с 2 a с
При малой a : a = .
Степень диссоциации слабого электролита зависит от концентрации раствора , уменьшаясь с ее ростом (см. рис. 22). Величина Кд,с , как и всякая константа равновесия, не должна зависеть от исходной концентрации слабого электролита, но на практике она является постоянной только для очень разбавленных растворов.
Рис. 22. Зависимость степени диссоциации слабого электролита a от его концентрации с
Рис. 23. Зависимость константы диссоциации и степени диссоциации слабого электролита от температуры
Степень диссоциации a , а следовательно и Кд,с , зависят также от температуры , зависимость проходит через максимум (см. рис. 23). Это можно объяснить влиянием двух противоположно направленных воздействий. С одной стороны, всякая диссоциация протекает с поглощением тепла, и, следовательно, при повышении температуры равновесие должно смещаться в сторону большей диссоциации. С другой стороны, при повышении температуры диэлектрическая проницаемость воды, служащей растворителем, уменьшается, а это способствует воссоединению ионов. Кд,с максимальна при той Т, при которой влияние второго фактора начинает преобладать. Обычно изменение Кд,с с повышением Т невелико.
Зависимость Кд,с от температуры описывается уравнением изобары Вант-Гоффа:
где D Ндис – теплота диссоциации. Работу диссоциации Адис определяют по уравнению изотермы Вант-Гоффа:
Теория электролитической диссоциации (Аррениус, Менделеев, Каблуков). Электролиты и неэлектролиты. Понятие о степени и константе диссоциации
Электролитическая диссоциация — процесс распада электролита на ионы при его растворении или плавлении.
Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Аррениус придерживался физической теории растворов, не учитывал взаимодействие электролита с водой и считал, что в растворах находятся свободные ионы. Русские химики И. А. Каблуков и В. А. Кистяковский применили для объяснения электролитической диссоциации химическую теорию растворов Д. И. Менделеева и доказали, что при растворении электролита происходит его химическое взаимодействие с водой, в результате которого электролит диссоциирует на ионы.
Классическая теория электролитической диссоциации основана на предположении о неполной диссоциации растворённого вещества, характеризуемой степенью диссоциации α, т. е. долей распавшихся молекул электролита. Динамическое равновесие между недиссоциированными молекулами и ионами описывается законом действующих масс .
Вещества, распадающиеся на ионы, называют электролитами. Электролиты – вещества с ионной или сильно ковалентной связью: кислоты, основания, соли. остальные вещества – неэлектролиты; к ним относятся вещества с неполярной или слабо полярной ковалентной связью; например, многие органические соединения.
Теория Электролитической диссоциации:
1. При растворении в воде электролиты распадаются на положительные ионы (катионы) и отрицательные ионы (анионы). ионы в растворе взаимодействуют с молекулами воды (гидратация). Процесс диссоциации является обратимым.
2. Под действием постоянного электрического тока катионы движутся по катоду, анионы – к аноду.
3. Степень диссоциации зависит от природы электролита и растворителя, концентрации электролита и температуры.
Степень диссоциации (а) – отношение числа молекул, распавшихся на ионы (N’) к общему числу растворенных молекул (N): а = N’/ N;
Сильный электролит – вещество, степень диссоциации которого больше 30%.. к сильным электролитам относят все соли , сильные кислоты, сильные основания.
Слабый электролит – вещество, степень диссоциации которого меньше 3%. к слабым электролитам относят слабые кислоты, слабые основания.
Степень диссоциации зависит от концентрации вещества в растворе, поэтому некоторые слабые электролиты при разбавлении могут стать сильными.
Константа диссоциации – константа равновесия электролитической диссоциации. она равна отношению произведений концентраций ионов, образующихся при диссоциации, к концентрации исходных частиц.
Не нашли то, что искали? Воспользуйтесь поиском:
Лучшие изречения: Для студента самое главное не сдать экзамен, а вовремя вспомнить про него. 9116 —  | 6863 —
| 6863 —  или читать все.
или читать все.
193.124.117.139 © studopedia.ru Не является автором материалов, которые размещены. Но предоставляет возможность бесплатного использования. Есть нарушение авторского права? Напишите нам | Обратная связь.
Отключите adBlock!
и обновите страницу (F5)
очень нужно
Теория электролитической диссоциации Аррениуса
Читайте также:
- A. Организм. Индивид. Личность. Ииндивидуальность. Теория развития индивидуальности Б.Г. Ананьева.
- I.4. Общая теория относительности (ОТО) и теория гравитации
- L Ситуативная теория (Т.Хилтон)
- XY» — Теория Д.макгрегора.
- Адсорбционная теория Штерна
- БИХЕВИОРИЗМ И ТЕОРИЯ СОЦИАЛЬНОГО НАУЧЕНИЯ
- Боровская теория атома.
- Бюрократическая теория организации
- В современной российской философии (как и в прежней советской философии) широко распространено материалистическое объяснение природы сознания,известное как теория отражения.
- Введение в менеджмент: теория и практика
- Взаимодействия. Концепция поля, гравитационное и электромагнитное поля. Волновая теория света
- Влияние температуры. Уравнение Аррениуса
Теории растворов
Равновесия в растворах электролитов
При всем многообразии растворов некоторые свойства присущи всем растворам, независимо от их природы.
Например, давление пара над раствором всегда ниже, чем над чистым растворителем, раствор всегда имеет более высокую температуру кипения и более низкую температуру замерзания по сравнению с чистым растворителем.
Направление процесса диффузии всегда – из области с большей концентрацией в область с меньшей.
И, наконец, присущее всем растворам явление осмоса: при наличии полупроницаемой мембраны наблюдается прохождение через нее молекул растворителя, приводящее к уменьшению концентрации более концентрированного раствора.
Кроме того, из курса физхимии известно понятие об идеальных и реальных растворах. Для идеальных растворов известны простые соотношения, связывающие их свойства с концентрацией. Такие соотношения справедливы лишь для очень разбавленных растворов. На самом деле физическая природа растворов весьма сложна, пэтому до настоящего времени теория растворов далека до завершения.
Существовало много теорий, опиисывющих закономерности поведения растворов. Одна из них – физическая, или газовая, теория создана Вант-Гоффом. С точки зрения этой теории частицы растворенного вещества взаимодействуют друг с другом и частицами растворителя только по законам упругого соударения, т.е. ведут себя как частицы идеального газа. При этом растворитель рассматривается лишь как среда, в которой распределены частицы растворенного вещества. Это очень упрощенная модель, но она позволила вывести некоторые важные количественные закономерности, справедливые, правда, лишь для разбавленных растворов.
В химической теории, разработанной Д.И.Менделеевым, предполагается, что частицы растворенного вещества могут взаимодействовать с растворителем, образуя с ним сложные соединения постоянного или переменного состава. Такие соединения были названы сольватами или сольватными комплексами, а процесс их образования – сольватацией (или в случае водных растворов – гидраты и гидратация соответственно).
В физической теории растворов было установлено, что свойства идеальных растворов являются простой функцией концентрации, выраженной через число молей растворенного вещества. Однако, существовал ряд растворов для которых эти зависимости давали неверный результат. Эти растворы обладали рядом особых свойств, одним из которых была электрическая проводимость. Впоследствии уже было установлено, что существуют 2 механизма прохождения электрического тока по проводникам – электронная проводимость и ионная. (Хотя такое деление не является вполне строгим. Предполагается, например, что металлы обладают электронной проводимостью, а соли в твердом, расплавленном или растворенном виде – ионной. Но, например, растворы щелочных металлов в жидком аммиаке обладают смешанной – электронной и ионной — проводимостью).
Вещества, растворы которых обладают преимущественно ионной растворимостью, стали называть электролитами. Помимо высокой проводимости электролиты обладают еще рядом особенностей, отличающих их растворы от других.
Первой количественной теорией растворов электролитов была теория шведского ученого Сванте Аррениуса, выдвинутая им в 1883-1887 гг. Дальнейшее свое развитие она получила в трудах Оствальда, Вальдена, Писаржевского и др.
Основные положения теории Аррениуса (физической теории слабых электролитов):
1.Электролиты способны при растворении в некоторых растворителях самопроизвольно распадаться на ионы – противоположно заряженные частицы. Этот распад был назван диссоциацией – отсюда и название теории.
2. Электролиты при растворении распадаются не полностью. Доля молекул, распавшихся на ионы, отвечает степени электролитической диссоциации и зависит в данном электролите при данной темепратуре от природы вещества и от его концентрации.
3. Силы взаимодействия между ионами отсутствуют, и растворы ведут себя подобно идеальным газовым системам. (Это положение авторами теории не высказывалось, но оно лежит в основе всех ее количественных соотношений).
Существовало много экспериментальных подтверждений теории Аррениуса.
1. Коллигативные свойства растворов электролитов (осмотическое давление, темп. замерзания и кипения) изменяются не так, как у неэлектролитов.
2. Постоянство величины теплоты нейтрализации различных кислот и оснований.
3. Постоянство числа Фарадея.
Теория Аррениуса, т.о., позволила вскрыть суть и механизмы реакций, протекающих в ионной форме в растворах. Кроме того, на ее основе были развиты следующие научные представления:
1. Теория кислот и оснований Бренстеда и Льюиса
3. Поянтие о буферных растворах
4. Теория индикаторов
5. Теории гидролиза и сольволиза
6. Теория комплексообразования в растворах
7. Понятие о ионном произведении и ПР малорастворимых соединений
8. Теории кислотно-основного кактализа
Однако, у теории были и неудачи, свидетельствующие о ее недостатках:
1. Степень диссоциации по теории должна быть постоянной величиной при заданных условиях вне зависимости от способа ее измерения и концентрации.
2. Эта величина не может быть больше 1 и меньше 0 в соответствии с ее физическим смыслом. Однако, было установлено, что степень диссоциации, измеренная с помощью электропроводности и по величине осмотического давления существенно различаются.
3. Константа диссоциации не должна зависеть от концентрации. Однако, это правило выполняется лишь для очень слабых электролитов.
Таким образом, основные недостатки теории связаны с количественными характеристиками:
1. Степень электролитической диссоциации не отвечает тому физическому смыслу, который вкладывается в нее теорией.
2. Константа диссоциации не является постоянной величиной.
3. (Самое важное) не указываются причины, вызывающие диссоциацию электролитов в растворах.
Все эти недостатки в конечном счете явились результатом неучтенного влияния взаимодействия между ионами в растворе.
Дата добавления: 2014-01-07 ; Просмотров: 2117 ; Нарушение авторских прав? ;
Нам важно ваше мнение! Был ли полезен опубликованный материал? Да | Нет
Кислоты и основания
Несмотря на то, что понятия «кислота» и «основание» широко используются для описания химических процессов, единого подхода к классификации веществ с точки зрения отнесения их к кислотам или основаниям нет. Существующие в настоящее время теории (ионная теория С. Аррениуса, протолитическая теория И. Бренстеда и Т. Лоури и электронная теория Г. Льюиса) имеют определенные ограничения и, таким образом, применимы лишь в частных случаях. Остановимся подробнее на каждой из этих теорий.
1. Теория Аррениуса.
В ионной теории Аррениуса понятия «кислота» и «основание» тесно связаны с процессом электролитической диссоциации:
Кислотой является электролит, диссоциирующий в растворах с образованием ионов Н + ;
Основанием является электролит, диссоциирующий в растворах с образованием ионов ОН ;
Амфолитом (амфотерным электролитом) является электролит, диссоциирующий в растворах с образованием как ионов Н + , так и ионов ОН .
H + MeOn n ⇄ Ме(ОН)n ⇄ Ме n + nОН
В соответствии с ионной теорией кислотами могут быть как нейтральные молекулы, так и ионы, например:
Аналогичные примеры можно привести и для оснований:
КОН  К + ОН
К + ОН
К амфолитам относят гидроксиды цинка, алюминия, хрома и некоторые другие, а также аминокислоты, белки, нуклеиновые кислоты.
В целом, кислотно-основное взаимодействие в растворе сводится к реакции нейтрализации:
H + ОН 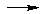 H2O
H2O
Однако, ряд экспериментальных данных показывает ограниченность ионной теории. Так, аммиак, органические амины, оксиды металлов типа Na2O, СаО, анионы слабых кислот и т.д. в отсутствии воды проявляют свойства типичных оснований, хотя не имеют в своем составе гидроксид-ионов.
С другой стороны, многие оксиды (SO2, SO3, Р2О5 и т.д.), галогениды, галогенангидриды кислот, не имея в своем составе ионов водорода, даже в отсутствии воды проявляют кислотные свойства, т.е. нейтрализуют основания.
Кроме того, поведение электролита в водном растворе и в неводной среде может быть противоположным.
Так, CH3COOH в воде является слабой кислотой:
а в жидком фтороводороде проявляет свойства основания:
Исследования подобных типов реакций и в особенности реакций, протекающих в неводных растворителях, привели к созданию более общих теорий кислот и оснований.
2. Теория Бренстеда и Лоури.
Дальнейшим развитием теории кислот и оснований явилась предложенная И. Бренстедом и Т. Лоури протолитическая (протонная) теория. В соответствии с этой теорией:
Кислотой называют любое вещество, молекулы (или ионы) которого способны отдавать протон, т.е. быть донором протона;
Основанием называют любое вещество, молекулы (или ионы) которого способны присоединять протон, т.е. быть акцептором протона;
Таким образом, понятие основания значительно расширяется, что подтверждается следующими реакциями:
ОН Н +  Н2О
Н2О
NH3 H + 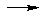 NH4 +
NH4 +
H2NNH3 + H + 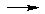 H3N + NH3 +
H3N + NH3 +
По теории И. Бренстеда и Т. Лоури кислота и основание составляют сопряженную пару и связаны равновесием:
КИСЛОТА ⇄ ПРОТОН ОСНОВАНИЕ
Поскольку реакция переноса протона (протолитическая реакция) обратима, причем в обратном процессе тоже передается протон, то продукты реакции являются друг по отношению к другу кислотой и основанием. Это можно записать в виде равновесного процесса:
где НА – кислота, В – основание, ВН + – кислота, сопряженная с основанием В, А – основание, сопряженное с кислотой НА.
HCl OH ⇄ Cl H2O,
HCl и H2O – кислоты, Cl и OH – соответствующие сопряженные с ними основания;
HSO4 и H3O + – кислоты, SO4 2 и H2O – основания;
NH4 + – кислота, NH2 – основание, а NH3 выступает в роли как кислоты (одна молекула), так и основания (другая молекула), т.е. демонстрирует признаки амфотерности – способности проявлять свойства кислоты и основания.
Такой способностью обладает и вода:
Здесь одна молекула Н2О присоединяет протон (основание), образуя сопряженную кислоту – ион гидроксония Н3О + , другая отдает протон (кислота), образуя сопряженное основание ОН . Этот процесс называется автопротолизом.
Из приведенных примеров видно, что в отличие от представлений Аррениуса, в теории Бренстеда и Лоури реакции кислот с основаниями не приводят к взаимной нейтрализации, а сопровождаются образованием новых кислот и оснований.
Необходимо также отметить, что протолитическая теория рассматривает понятия «кислота» и «основание» не как свойство, но как функцию, которую выполняет рассматриваемое соединение в протолитической реакции. Одно и то же соединение может в одних условиях реагировать как кислота, в других – как основание. Так, в водном растворе СН3СООН проявляет свойства кислоты, а в 100%-й H2SO4 – основания.
Однако, несмотря на свои достоинства, протолитическая теория, как и теория Аррениуса, не применима к веществам, не содержащим атомов водорода, но, в тоже время, проявляющим функцию кислоты: галогенидам бора, алюминия, кремния, олова.
Электролитическая диссоциация: сущность теории С. Аррениуса
 Своё начало теория электролитов берёт ещё в первой половине XIX века, когда М. Фарадей провёл свои знаменитые опыты с растворами поваренной соли. Он установил, что абсолютно чистая вода очень плохо проводит электрический ток, но стоит добавить в неё несколько кристаллов соли, и проводимость тут же возрастает. Уже тогда родилось предположение, что соль распадается в воде на некие частицы, которые способны проводить электрический ток, однако, полноценная теория, описывающая все эти процессы в растворах, появилась гораздо позже.
Своё начало теория электролитов берёт ещё в первой половине XIX века, когда М. Фарадей провёл свои знаменитые опыты с растворами поваренной соли. Он установил, что абсолютно чистая вода очень плохо проводит электрический ток, но стоит добавить в неё несколько кристаллов соли, и проводимость тут же возрастает. Уже тогда родилось предположение, что соль распадается в воде на некие частицы, которые способны проводить электрический ток, однако, полноценная теория, описывающая все эти процессы в растворах, появилась гораздо позже.
Теория электролитической диссоциации
 Теория, основоположником которой явился Сванте Аррениус в период 1883—1887 гг., базируется на идее, что при попадании молекул растворимого вещества (электролита) в полярную или неполярную жидкость происходит их диссоциация на ионы. Электролитами называются соединения, которые в растворе самопроизвольно распадаются на ионы, способные к самостоятельному существованию. Количество образующихся ионов, их строение и величина заряда зависят только от природы диссоциировавшей молекулы.
Теория, основоположником которой явился Сванте Аррениус в период 1883—1887 гг., базируется на идее, что при попадании молекул растворимого вещества (электролита) в полярную или неполярную жидкость происходит их диссоциация на ионы. Электролитами называются соединения, которые в растворе самопроизвольно распадаются на ионы, способные к самостоятельному существованию. Количество образующихся ионов, их строение и величина заряда зависят только от природы диссоциировавшей молекулы.
 Для использования теории в описании свойств растворения используется ряд допущений, а именно: предполагается, что диссоциация является неполной, ионы (их электронные оболочки) не реагируют друг с другом, а их поведение можно описать законом действующих масс в идеальных условиях. Если рассмотреть теоретическую систему, где электролит КА находится в фазовом равновесии с продуктами своей диссоциации — катионом К+ и анионом А-, то согласно закону действующих масс можно составить уравнение реакции диссоциации:
Для использования теории в описании свойств растворения используется ряд допущений, а именно: предполагается, что диссоциация является неполной, ионы (их электронные оболочки) не реагируют друг с другом, а их поведение можно описать законом действующих масс в идеальных условиях. Если рассмотреть теоретическую систему, где электролит КА находится в фазовом равновесии с продуктами своей диссоциации — катионом К+ и анионом А-, то согласно закону действующих масс можно составить уравнение реакции диссоциации:
Константа равновесия, записанная, через концентрации веществ при изотермических условиях будет иметь следующее значение:
Кд = [K+] x [A-] / [KA] (2)
 В этом случае (в уравнении 2), константа равновесия Кд, будет являться не чем иным, как константой диссоциации, значения [KA], [K+], [A-] в правой части — это равновесные концентрации электролита и его продуктов диссоциации.
В этом случае (в уравнении 2), константа равновесия Кд, будет являться не чем иным, как константой диссоциации, значения [KA], [K+], [A-] в правой части — это равновесные концентрации электролита и его продуктов диссоциации.
Учитывая допущение теории Аррениуса, которые были применены автором, в частности, о неполноте диссоциации, вводится понятие степени диссоциации — α. Таким образом, если выразить концентрацию раствора С (моль/л), то на литр раствора приходится αС моль электролита (КА), а равновесная его концентрация может быть выражена, как (1-α)С моль/л. Из уравнения реакции (1) очевидно, что на αС моль электролита (КА) образуется такое же количество ионов К+ и А-. Если подставить все эти величины в уравнение (2) и провести ряд упрощений, то получим формулу константы диссоциации (степень диссоциации формула):
 Это уравнение позволяет количественно определить величину степени электролитической диссоциации в разных растворах.
Это уравнение позволяет количественно определить величину степени электролитической диссоциации в разных растворах.
Теория Аррениуса дала развитие множеству научных направлений в химии: с её помощью были созданы первые теории кислот и оснований, были даны объяснения физико-химическим процессам в гомогенных системах. Тем не менее, она не лишена недостатков, которые в основном относятся к тому факту, что теория не учитывает межионные взаимодействия.
Классификация электролитов с примерами
Электролиты классифицируют на слабые и сильные, периодически выделяя группу электролитов средней силы. Сильные электролиты характеризуются тем, что распадаются в растворе полностью. Как правило — это сильные минеральные кислоты, например:
- Азотная кислота — HNO3.
- Хлороводородная кислота — HCl.
- Хлорная кислота — HClO4.
- Ортофосфорная кислота — H3PO4.
Сильными электролитами могут быть основания, например:
 Основная масса сильных электролитов — это подавляющее большинство солей (NaCl, Na2SO4, Ca (NO3)2, CH3COONa, хлориды, сульфиды).
Основная масса сильных электролитов — это подавляющее большинство солей (NaCl, Na2SO4, Ca (NO3)2, CH3COONa, хлориды, сульфиды).
Слабые электролиты, напротив, в растворах гидратируют частично. К этой группе следует относить неорганические кислоты (H2CO3, H3BO3, H3AsO4), слабые основания (аммоний), некоторые соли (HgCl2), органические кислоты (CH3COOH, C6H5COOH), фенолы и амины. В неводных растворах одни и те же соединения могут являться и сильными и слабыми электролитами, таким образом, зависят от природы растворителя.
Диссоциация кислот, оснований и солей
Закономерности для кислот
При электрической диссоциации кислот в водных растворах обязательно в качестве катионов образуются положительно заряженные ионы водорода (Н+):
Если кислота многоосновная (например: уравнение диссоциации H2SO4), то диссоциация происходит последовательно, за каждый раз отщепляя один ион водорода:
H2SO4 → H + + HSO4- первая ступень — гидросульфат ион
HSO4- → H + + SO4- вторая ступень — сульфат ион
Процесс для многоосновной кислоты, как правило, протекает максимально по первой ступени, степень диссоциации последующих намного меньше.
Характеристика процесса для щелочей
При диссоциации щелочей в водных растворах обязательно образуется отрицательно заряженный гидроксил ион (ОН-):
Процесс для многокислотных оснований (пример — механизм диссоциации гидроксида магния) протекает многоступенчато аналогично многоосновным кислотам:
Mg (OH)2 → OH- + Mg (OH)+ первая ступень
Mg (OH)+ → OH- + Mg2+ вторая ступень
Существуют также случаи, когда в процессе диссоциации могут образовываться и катионы водорода, и гидроксил-анионы (при диссоциации амфолитов или амфотерных соединений, например, Zn, Al):
2OH- + Zn2+ + 2H2O ←→ Zn (OH)2 + H2O ←→ [Zn (OH)4]2- + 2H+
Правила протекания для кислых и основных солей
Для кислых солей, основная закономерность заключается в следующем — сначала диссоциируют катионы (положительно заряженные металлы), а только потом катионы водорода:
KHSO4 → K+ + HSO4- первая ступень
HSO4 — → H+ + SO4- вторая ступень
У основных солей, в первую очередь, переходят в раствор остатки кислоты, а уже затем гидроксил-ион:
BaOHCl → Cl- + Ba (OH)+ первая ступень
Ba (OH)+ → OH- + Ba2+ вторая ступень
Водородный показатель
Определение, сущность и значение
Процессы диссоциации могут протекать не только для растворенных веществ, но и растворителя. Так, вода является сама со себе слабым электролитом и для неё характерна диссоциация в очень незначительной степени. Уравнение процесса можно записать следующим образом:
Одна молекула воды диссоциирует на положительно заряженные ионы водорода и отрицательно заряженные анионы гидроксония. Именно концентрация этих ионов определяет уровень кислотности раствора — чем больше ионов гидроксония, тем более кислый раствор.
Концентрация ионов гидроксония в реальных растворах, как правило, очень мала (например: 5×10−6 г/л) и поэтому для удобства, это значение логарифмируют, а чтобы получить положительное значение, берут с обратным знаком. Кратко сформулируем строгое определение понятия «водородный показатель» или рН.
рН (водородный показатель) — это отрицательный натуральный логарифм концентрации ионов гидроксония, отражающий кислотность раствора.
Значения водородного показателя принято оценивать по шкале значений от 0 до 14, где 0 — наиболее кислый раствор, а 14 — наиболее щелочной. Нейтральным раствором (соответствующим рН чистой воды) считается раствор со значением 7. Для примера приводим несколько типичных растворов, имеющих характерные значения водородного показателя:
| Значение рН | Раствор |
| 11 | Нашатырный спирт |
| 9,5 | Гидроксид кальция |
| 8,0 | 30% раствор поваренной соли |
| 7,4 | Плазма крови |
| 7,0 | Деионизированная вода |
| 6,5 | Молоко |
| 5,5 | Кофе |
| 2,8 | Уксус (раствор 5% концентрации) |
| 0,1 | Хлорная кислота (65%) |
Значительно реже прибегают к использованию еще одного показателя — рОН. По своему смыслу он абсолютно аналогичен водородному показателю, за исключением того, что за основу берётся концентрация гидроксил-ионов.
Теория электролитической диссоциации Аррениуса.
Для объяснения особенностей водных растворов электролитов шведским химиком С. Аррениусом в 1887 г. была предложена теория электролитической диссоциации. Основные положения теории следующие:
1. Электролиты при растворении в воде распадаются на ионы – положительные и отрицательные. Ионы находятся в более устойчивых электронных состояниях, чем атомы. Среди таких ионов встречаются простые, например, Na + , Mg 2+ , Al 3+ и сложные, состоящие из нескольких атомов, например, NO3 — , SO4 2- , PO4 3- . В растворе ионы беспорядочно передвигаются в различных направлениях.
2. Под действием электрического тока ионы приобретают направленное движение: положительно заряженные ионы движутся к катоду, отрицательно заряженные – к аноду. Первые называются катионами, а вторые анионами.
3. Диссоциация – обратимый процесс. Одновременно с распадом молекул на ионы протекает обратный процесс – соединения ионов в молекулу.
Поэтому в уравнениях электролитической диссоциации стоит не знак равенства, а знак обратимости ↔. Например, уравнение диссоциации молекулы электролита КА на катион К + и анион А — записывается в виде
КА 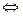 К + + А — (1).
К + + А — (1).
Теория электролитической диссоциации полностью согласуется с атомно-молекулярным учением и теорией строения атома.
Степень диссоциации электролитов
Под степенью диссоциации электролита понимается отношение числа диссоциированных на ионы молекул n к общему числу молекул растворенного электролита N, то есть
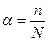 (2).
(2).
В зависимости от степени диссоциации различают слабые и сильные электролиты. Сильные электролиты при больших концентрациях диссоциированы более чем на 1/2. Степень диссоциации слабых электролитов очень мала по сравнению с 1. Сильные электролиты – это большинство нейтральных солей, сильные кислоты (НСl, HClO4, H2SO4), сильные основания (NaOH, KOH). Слабые электролиты – это большинство органических кислот, неорганические слабые кислоты и слабые основания, некоторые нейтральные соли CdCl2, Fe(CH3COO)3. Особенно слабыми электролитами являются вода, сероводород, синильная и борная кислоты.
Между сильными и слабыми электролитами существует переходная группа, которую образуют соли тяжелых металлов, а также некоторые сильные органические кислоты: лимонная, щавелевая, муравьиная.
Степень диссоциации зависит от природы электролита и растворителя, а также от концентрации электролита. С уменьшением концентрации степень диссоциации увеличивается, а при сильном разведении раствора, а→1, и различия между сильно и слабодиссоциирующими электролитами сглаживаются.
Степень диссоциации зависит и от диэлектрической проницаемости растворителя ε. Чем больше диэлектрическая проницаемость, тем сильнее диссоциирует электролит. В воде электролитическая диссоциация может быть сильной, а в ацетоне и в особенности бензоле – слабой. Диэлектрическая проницаемость ε воды, ацетона и бензола соответственно равна 80, 21 и 2,3. Эта закономерность, установленная Нернстом и Томсоном в 1893 г., объясняется тем, что, согласно закону Кулона, сила притяжения между разноименно заряженными ионами обратно пропорциональна диэлектрической проницаемости ε.
Дата добавления: 2017-02-13 ; просмотров: 2825 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Учебное пособие для модульно-рейтинговой технологии обучения Бийск
Теория электролитической диссоциации С. Аррениуса содержит следующие положения:
1) при растворении в воде (или расплавлении) электролиты распадаются на положительно и отрицательно заряженные ионы (подвергаются электролитической диссоциации);
2) под действием электрического тока катионы (+) двигаются к катоду (-), а анионы (-) – к аноду (+);
3) электролитическая диссоциация – процесс обратимый (обратная реакция называется моляризацией);
4) степень электролитической диссоциации α (Приложение Г) зависит от природы электролита и растворителя, температуры и концентрации. Она показывает отношение числа молекул, распавшихся на ионы ( n ), к общему числу молекул, введенных в раствор ( N ):
 . (2.37)
. (2.37)
Степень диссоциации слабого электролита можно связать с изотоническим коэффициентом. Будем считать, что из N молекул электролита продиссоциировало n молекул, образовав νn ионов ( ν – число ионов, на которое диссоциирует молекула). Поскольку изотонический коэффициент показывает, во сколько раз общее число молекул и ионов в растворе больше числа молекул до диссоциации, получаем:
 , (2.38)
, (2.38)
 . (2.39)
. (2.39)
Соотношение (2.39) дает возможность, экспериментально определив изотонический коэффициент раствора, рассчитать степень диссоциации слабого электролита.
Однако теория С. Аррениуса не учитывала всей сложности явлений в растворах. В частности, она рассматривала ионы как свободные, независимые от молекул растворителя частицы. Теории С. Аррениуса противостояла химическая, или гидратная, теория растворов Д.И. Менделеева, в основе которой лежало представление о взаимодействии растворенного вещества с растворителем. Кажущееся противоречие обеих теорий было устранено предположением о гидратации ионов (И.А. Каблуков).
Сольватация – взаимодействие между молекулами и ионами растворяемого вещества и молекулами растворителя, состоит из нескольких стадий:
а) Молекулярная диссоциация и образование сольватов (рисунок 2.5).

Рисунок 2.5 – Процесс образования сольватов
 ,
,
где АВ – молекулы растворяемого вещества;
S – молекулы растворителя.
б) Ионизация и электролитическая диссоциация (рисунок 2.6).

Рисунок 2.6 – Электролитическая диссоциация
 .
.
Электролитическая диссоциация − это процесс распада молекул веществ на ионы под действием полярных молекул растворителя, а также при их плавлении.
Если процесс сольватации останавливается на стадии (a), то образуется раствор неэлектролита (подчиняется законам Рауля, принципу Вант-Гоффа) – раствор сахара в воде.
Если процесс протекает до стадии (б ) , то система является раствором электролита: растворы щелочей, солей, неорганических кислот в воде.
Механизм электролитической диссоциации ионных веществ можно представить следующим образом.
При растворении соединений с ионными связями (например, NaCl) процесс гидратации начинается с ориентации диполей воды вокруг всех выступов и граней кристаллов соли.
Ориентируясь вокруг ионов кристаллической решетки, молекулы воды образуют с ними либо водородные, либо донорно-акцепторные связи. При этом процессе выделяется большое количество энергии, которая называется энергией гидратации.
Энергия гидратации, величина которой сравнима с энергией кристаллической решетки, идет на разрушение кристаллической решетки. При этом гидратированные ионы слой за слоем переходят в растворитель и, перемешиваясь с его молекулами, образуют раствор.
Механизм электролитической диссоциации полярных веществ можно представить следующим образом.
Аналогично диссоциируют и вещества, молекулы которых образованы по типу полярной ковалентной связи. Вокруг каждой полярной молекулы вещества (например, HCl) определенным образом ориентируются диполи воды. В результате взаимодействия с диполями воды полярная молекула еще больше поляризуется и превращается в ионную, далее уже легко образуются свободные гидратированные ионы.
2.7.2 Вопросы для самоконтроля
Что такое сольваты?
Что называется электролитической диссоциацией, или ионизацией?
Кто и когда предложил теорию электролитической диссоциации?
Сформулируйте основные положения теории электролитической диссоциации.
Какая химическая связь существует в молекулах электролитов?
Как происходит диссоциация электролитов с ионной связью? Покажите схематически на примере.
Как происходит диссоциация электролитов с полярной связью? Покажите на примере.
Что называется степенью диссоциации?
Каким соотношением связаны между собой изотонический коэффициент и степень электролитической диссоциации?
2.8 Теории кислот и оснований
2.8.1 История развития представлений о кислотах
Множество химических реакций может быть рассмотрено с позиций кислотно-основных взаимодействий. Такие вопросы, как трактовка механизмов реакций, кислотный и основной катализ, влияние различных факторов на ход процесса, в основном базируются на учете кислотно-основных взаимодействий. Овладеть оценкой кислотно-основных взаимодействий − это значит понять сущность химической реакции и иметь возможность управлять ею.
Представления о кислотах и основаниях существуют более трех столетий, но до сих пор нет единого определения этих понятий. Рассмотрим кратко эволюцию взглядов на природу кислот и оснований. Термины «кислоты» и «основания» сформировались еще в XVII веке, однако их содержание неоднократно пересматривалось и уточнялось. Лишь в конце прошлого века после появления теории электролитической диссоциации (С. Аррениус, 1887 г.) сформировалась первая научная ионная теория кислот и оснований (Д. Оствальд, С. Аррениус).
Важной вехой в развитии взглядов на кислоты и основания явилось сформулированное А. Ганчем в 1917–1927 годах понятие об амфотерности − способности некоторых соединений проявлять как кислотные, так и основные свойства в зависимости от условий и природы реагентов, участвующих в кислотно-основном взаимодействии. И, как оказалось позже, таких соединений (проявляющих амфотерность) − подавляющее большинство. В зависимости от природы партнера по взаимодействию явная кислота может выступать в роли основания и наоборот.
Доминирующие в настоящее время протонная (И. Бренстед и Т. Лаури) и электронная (Г. Льюис) теории кислот и оснований были предложены одновременно в 1923 году.
Академиком АН Казахской ССР физиком-химиком М.И. Усановичем была предпринята попытка объединить электронную и протонную теории. В 1938 г. М.И. Усанович разработал оригинальную теорию кислот и оснований, положив в основу представление, что водород не является универсальным носителем кислотных свойств. Он отнес к кислотам все вещества, образующие соли при взаимодействии с основаниями. Все окислительно-восстановительные и протолитические реакции оказывались частным случаем кислотно-основных взаимодействий. Кроме того, М.И. Усанович считал, что амфотерность присуща всем полярным соединениям.
В современной химии используются две теории кислот и оснований: теория Бренстеда − Лаури и теория Льюиса − Усановича. Более общие определения кислот и оснований предложены в теории Льюиса – Усановича.
2.8.2 Кислоты и основания в теории С. Аррениуса
С. Аррениус объяснял появление ионов в водном растворе (или расплаве) распадом растворенных веществ − электролитов .
Аррениус из всех ионов выделил ионы H + и OH — как продукты автодиссоциации воды :
 .
.
Затем он высказал утверждение, что кислотой называют электролит, диссоциирующий в растворах с образованием ионов Н + ; основанием называют электролит, диссоциирующий в воде с образованием гидроксид-ионов ОН  :
:
кислоты:  ;
;
основания:  .
.
Кислоты и основания, для которых степень распада на ионы (степень электролитической диссоциации) имеет значение, близкое к единице, были названы С. Аррениусом сильными ( HNO 3 , NaOH ), а все остальные – слабыми ( HNO 2 , NH 3 . H 2 O ).
Слабые кислоты можно было сравнить по силе, используя значения констант диссоциации кислот K ДК , а слабые основания − по значениям констант диссоциации оснований K ДО . Чем выше значение констант диссоциации K Д , тем более сильным считался слабый электролит-кислота или слабый электролит-основание.
 ,
,
 .
.
 ,
,
 .
.
Сильные же кислоты и основания с точки зрения теории кислот и оснований, предложенной С. Аррениусом, никак не характеризовались количественно , их сила просто считалась наибольшей.
2.8.3 Протонная теория кислот и оснований
Протонная теория Брёнстеда − Лаури применима лишь к протонсодержащим или протонприсоединяющим веществам. Согласно этой теории кислотой называется вещество, способное быть донором протонов, а основанием – вещество, которое может присоединить (акцептировать) протон:
 .
.
По определению, H А n – кислота, An — – основание, сопряженное с этой кислотой. Любой кислоте соответствует сопряженное с ней основание:
 .
.
Любое кислотно-основное равновесие включает взаимодействие двух пар кислот и оснований. Например, в реакции

кислота 1 основание 2 основание 1 кислота 2
сопряженными парами будут HNO 3 / NO  и NH 4 + / NH 3 .
и NH 4 + / NH 3 .
CH 3 COOH + NH 3 ↔ CH 3 COO — + NH 4 + .
кислота основание сопряженное сопряженная
Кислоты и основания как вещества, теряющие и приобретающие протоны, называются протолитами . Передача протона от кислоты к основанию именуется протолизом , а химическая реакция между ними – протолитической реакцией .
В протолитических реакциях все вещества, будь то реагенты или продукты, проявляют кислотные или основные свойства. Протонная теория рассматривает каждое вещество в конкретной ситуации, исходя из выполняемой этим веществом химической функции ( донора или акцептора протонов ).
Протонодонорная и протоноакцепторная способность веществ (их кислотность и основность ) определяется сродством к протону, т.е. тепловым эффектом реакции присоединения протона:
NH 3(г) + H + (г) ↔NH 4 + (г) , Q = 868 кДж;
H 2 O (г) + H + (г) ↔H 3 O + (г) , Q = 689 кДж.
Здесь NH 3 – более сильный акцептор протона и более сильное основание, чем H 2 O , а NH 4 + , наоборот, более слабый донор протона и более слабая кислота, чем H 3 O + . В ряду жидких веществ:
NH 3 , H 2 O , C 2 H 5 OH , HCN , H 2 S , CH 3 COOH , HNO 3 , H 2 SO 4 , HF , HClO 4
сродство к протону убывает от NH 3 к HClO 4 .
Встречаются протолиты либо с однозначной функцией (кислоты, основания), либо с двойственной функцией ( амфолиты ). Амфолитом называют электролит, способный как отдавать, так и принимать протоны. Например, взаимодействие двух амфолитов:
H 2 O (г) + HF (г) ↔OH — (г) + H 2 F + (г) ,
кислота + основание ↔ основание + кислота
H 2 O (г) + HF (г) ↔H 3 O + (г) + F — (г).
основание + кислота ↔ кислота + основание
Многие гидроанионы, например HCO 3 — , являются в водном растворе амфолитами :
HCO 3 — + H 2 O ↔CO 3 2- + H 3 O + ,
HCO 3 — + H 2 O ↔H 2 CO 3 + OH — .
Амфолитами по отношению к воде являются также амфотерные гидроксиды , такие как Zn(OH) 2 , Be(OH) 2 , Al(OH) 3 , Cr(OH) 3 и другие. В водном растворе они гидратируются − образуют гидроксоаквакомплексы типа [Zn(H 2 O) 2 (OH) 2 ] и [Al(H 2 O) 3 (OH) 3 ].
2.8.4 Электронная теория Льюиса
В определенных условиях многие вещества могут вести себя как кислота или как основание. Эти два понятия неразделимы, а потому правильнее говорить о кислотно-основных свойствах данного вещества. В 1923 г. американский физик-химик Г. Н. Льюис сформулировал одну из основных современных теорий кислот и оснований – электронную.
Электронная теория Льюиса допускает, что участие в кислотно-основном равновесии протона необязательно, поэтому ее называют апротонной . Согласно апротонной (электронной) теории, кислотой называется вещество, способное присоединять электронную пару, а основанием – вещество, способное отдавать электронную пару:
 .
.
При взаимодействии донора электронной пары NF 3 (основание) и акцептора электронной пары BF 3 (кислота) образуется более устойчивое электронное окружение (октет) за счет донорно-акцепторной связи. Ни кислота, ни основание протонов не содержат.
Например, в реакции:
AlF 3 + :NH 3 = F 3 Al:NH 3 .
Алюминий помещает неподеленную электронную пару атома азота на свою вакантную электронную орбиталь. Интересен пример из органической химии, демонстрирующий механизм действия кислоты Льюиса (AlBr 3 ) в реакции бензола с бромом. Поляризующая способность молекулы бензола по отношению к брому оказывается недостаточной для успешного протекания реакции. На помощь и приходит молекула бромида алюминия, которая по отношению к брому обладает более сильной поляризующей способностью:
AlBr 3 + Br 2 = Br + [AlBr 4 — ] .
В данном примере неподеленная пара одного из атомов брома помещается на вакантную электронную орбиталь атома алюминия в бромиде алюминия.
Электронная теория Льюиса расширяет границы веществ, проявляющих кислотно-основные свойства, включая в себя протонотдающие и протонприсоединяющие системы.
Известны следующие типы кислот и оснований Льюиса:
а) протон и катионы металлов. Сила этих кислот возрастает с увеличением заряда и электроотрицательности металла; следовательно, если металл имеет несколько состояний окисления, самой сильной кислотой будет катион с наиболее высокой степенью окисления ( Sb 5+ , Sn 4+ , Fe 3+ );
б) наиболее распространенными кислотами Льюиса являются молекулы, центральный атом которых имеет одну, две или несколько вакантных орбиталей, например, соединения элементов второй и третьей групп периодической системы ( BX 3 , AlX 3 , BeX 2 , ZnX 2 );
в) ненасыщенные соединения. Молекулы с двойными, тройными связями реагируют с кислотами Льюиса как основания.
Считают, что наиболее сильной из известных кислот Льюиса является SbF 5 .
2.8.5 Кислотно-основные свойства соединений
В периоде сила кислородсодержащей кислоты растет с увеличением заряда и с уменьшением радиуса иона кислотообразующего элемента:
 .
.
Для одного и того же элемента константа диссоциации различных кислот возрастает по мере увеличения степени окисления кислотообразующего элемента примерно на пять порядков каждый раз.
 ,
,
 .
.
В пределах одной группы элементов сила кислоты уменьшается по мере увеличения радиуса кислотообразующего элемента:
 .
.
Сила же некислородных кислот возрастает в ряду однотипных соединений при переходе вниз по подгруппе кислотообразующих элементов:
 .
.
Для многоосновных кислот способность к депротонизации уменьшается по мере увеличения отрицательного заряда аниона:
 .
.
Для кислот характерны следующие общие свойства:
а) способность взаимодействовать с основаниями с образованием солей;
б) способность взаимодействовать с некоторыми металлами с выделением водорода;
в) способность изменять цвет индикаторов, в частности, вызывать красную окраску лакмуса;
Водные растворы оснований обладают следующими общими свойствами:
а) способностью взаимодействовать с кислотами с образованием солей;
б) способностью изменять цвет индикаторов иначе, чем для кислот (например, они вызывают синюю окраску лакмуса);
в) своеобразным мыльным вкусом.
Основные свойства гидроксидов одного и того же элемента усиливаются с уменьшением валентности элемента. Так, основные свойства дигидроксида железа Fe(OH) 2 выражены сильнее, чем у тригидроксида Fe(OH) 3 . Это утверждение равносильно тому, что кислотные свойства Fe(OH) 3 проявляются сильнее, чем у Fe(OH) 2 .
2.8.6 Вопросы для самоконтроля
1. Какова история развития представлений о кислотах и основаниях?
2. Сформулируйте понятие кислоты и основания с позиции теории С. Аррениуса.
3. Каковы основные положения протонной теории Бренстеда − Лаури?
4. Что собой представляют амфолиты?
5. Приведите примеры кислот и оснований на основе электронной теории Льюиса.
6. Какие существуют закономерности изменения кислотно-основных свойств соединений?
- http://spargalki.ru/himia/102-electrohimiya.html?start=16
- http://studopedia.ru/4_177063_teoriya-elektroliticheskoy-dissotsiatsii-arrenius-mendeleev-kablukov-elektroliti-i-neelektroliti-ponyatie-o-stepeni-i-konstante-dissotsiatsii.html
- http://studopedia.su/9_52216_teoriya-elektroliticheskoy-dissotsiatsii-arreniusa.html
- http://studfiles.net/preview/467580/page:4/
- http://1001student.ru/himiya/elektroliticheskaya-dissotsiatsiya-sushhnost-teorii-s-arreniusa.html
- http://poznayka.org/s85689t1.html
- http://refdb.ru/look/2300430-p5.html