
Значение растворов для химии невозможно переоценить. Огромное количество химических реакций происходит в растворах, а растворимость — одно из важнейших свойств вещества. Растворы изучает физическая химия. Раствор — это гомогенная, то есть однородная смесь переменного состава, которая состоит из растворителя, растворенного вещества и продуктов, получившихся в результате их взаимодействия. При этом каждый из компонентов распределяется в массе другого в виде молекул, ионов или атомов.
Под переменным составом раствора понимается следующее: соотношение веществ, смешанных друг с другом, способно в определенных пределах непрерывно изменяться. Раствор соли можно упаривать или разбавлять водой, но жидкости, полученные при этом, все равно будут называться растворами соли.
Растворитель отличается от растворенного вещества тем, что его агрегатное состояние при образовании раствора не изменяется. Обычно речь идет о растворе твердого вещества в жидкости, тогда жидкость и есть растворитель. Если же смешивается газ с газом, жидкость с жидкостью, твердое вещество с твердым, то растворителем является тот компонент, количество которого преобладает.
Образование раствора связано с интенсивностью межатомного, межмолекулярного или межионного взаимодействия частиц разных веществ. Процесс взаимодействия растворяемого вещества с водой называется гидратацией, что является частым случаем сольватации — взаимодействия частиц растворенного вещества с частицами растворителя. При гидратации образуются гидраты. Кристаллогидратами называются кристаллы, в состав которых входят молекулы воды такая вода носит название кристаллизационной. Образование водородных и других связей энергетически выгодно, и потому гидратация сопровождается выделением энергии. Ее часть идет на разрушение кристаллической решетки, а избыток выделяется в виде тепла. Так, при растворении твердого гидроксида натрия NaOH раствор сильно разогревается. Но если для разрушения кристаллической решетки расходуется больше энергии, чем выделяется при образовании гидратов, то раствор сильно охладится. Так происходит, например, при растворении в воде твердого нитрата аммония NH4N03.
Таким образом, вещество в растворе находится в новом состоянии — в виде гидратов. Значит, растворение не только физический, но и химический процесс.
В быту раствором называют только жидкое вещество, например раствор соли в воде или золота в ртути. Но в химической практике растворы — это гомогенные системы, в которых растворитель бывает жидким, твердым или газообразным. Смесь цемента с водой и песком тоже называют раствором, хотя с точки зрения химии это раствором не является.
Физическая и химическая теории растворов.
Растворы являются сложными системами, в которых имеют место различные виды взаимодействия между частицами (Ван-дер-Ваальсовы, электростатические и т.д.).
Существуют две точки зрения на природу растворения и растворов. Согласно физической точке зрения, растворение является чисто физическим процессом (разрушение кристаллической решетки при растворении твердых тел). Растворы при этом рассматриваются как молекулярные смеси нескольких веществ, не взаимодействующих химически. Противоположные представления были развиты Д. И. Менделеевым, который считал растворение химическим процессом, а растворы рассматривал как непрочные соединения компонентов раствора, находящихся в состоянии частичной диссоциации и отличающихся от обычных соединений переменным составом.
В настоящее время используются представления обеих теорий и доминирующая роль физической или химической компонент, в процессе растворения, определяется свойствами растворителя и растворенного вещества (системы).
Закон электронейтральности
При диссоциации молекул, число положительных и отрицательных ионов определяется стехиометрическими индексами в формуле молекулы. Электролиты, в которых ионы обладают одинаковым зарядом катиона и аниона, например, 1-1-электролит KCl 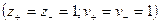 или 2-2-электролит
или 2-2-электролит 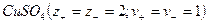 , распадаются на два иона — называются симметричные или симметричными. Электролиты, в которых ионы обладают неодинаковым зарядом катиона и аниона, например, 1-2-электролит
, распадаются на два иона — называются симметричные или симметричными. Электролиты, в которых ионы обладают неодинаковым зарядом катиона и аниона, например, 1-2-электролит 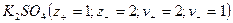 или 3-1–электролит
или 3-1–электролит 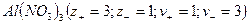 , называются несимметричными. Для любого типа электролита в элементарном объеме сумма зарядов анионов и катионов всегда одинакова (закон электронейтральности) :
, называются несимметричными. Для любого типа электролита в элементарном объеме сумма зарядов анионов и катионов всегда одинакова (закон электронейтральности) :
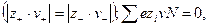 (32.1)
(32.1)
Где 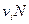 число частиц в растворе.
число частиц в растворе.
Степень диссоциации, изотонический коэффициент
Количественно диссоциация характеризуется степенью диссоциации 
 . (32.2)
. (32.2)
Величина 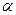 может изменяться от нуля (диссоциация отсутствует) до единицы (в растворе присутствуют только ионы). У сильных
может изменяться от нуля (диссоциация отсутствует) до единицы (в растворе присутствуют только ионы). У сильных  электролитов
электролитов 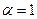 , у слабых —
, у слабых — 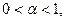 у неэлектролитов
у неэлектролитов 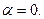
Изотонический коэффициент Вант-Гоффа i характеризует во сколько раз изменилось общее число частиц в растворе в результате диссоциации:
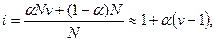
где числитель — общее число вещества в растворе: распавшихся на ионы 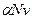 и оставшихся непродиссоциированными
и оставшихся непродиссоциированными 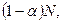 а знаменатель — число молекул, введенных в раствор.
а знаменатель — число молекул, введенных в раствор.
Для сильных электролитов изотонический коэффициент теоретически должен быть равен числу ионов, на которые распадается молекула при диссоциации: при 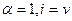 (например, для
(например, для  и
и 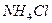 v=2, для
v=2, для 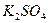 и
и 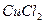 v=3, для
v=3, для  v=4 и т.д.). Однако обычно экспериментальные величины i
v=4 и т.д.). Однако обычно экспериментальные величины i
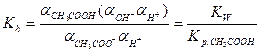
Как следует из приведенного выражения, константа гидролиза обратно пропорциональна константе диссоциации слабого электролита, участвующего в образовании соли (если в образовании соли участвуют два слабых электролита, то  обратно пропорциональна произведению их констант диссоциации).
обратно пропорциональна произведению их констант диссоциации).
Степень гидролиза является величиной аналогичной степени диссоциации.
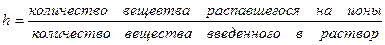
Уравнение, связывающую константу гидролиза со степенью гидролиза, по форме аналогично уравнению (32.3):
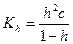
где h — число частиц введенных в раствор.
При повышении температуры степень диссоциации воды сильно увеличивается, тогда как у большинства других электролитов она изменяется незначительно. Вследствие этого степень гидролиза водных растворов при повышении температуры увеличивается.
Буферные растворы
В природе и практической деятельности многие реакции протекают при определенном значении pH, которое должно быть постоянным и не зависеть от разведения, изменения состава раствора, добавления кислоты или щелочи и т.д. Такими свойствами обладают буферные растворы, содержащие слабую кислоту и соль, образованную этой кислотой и сильным основанием (например, ацетатный буфер 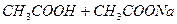 ), или слабое основание и соль, образованную сильной кислотой и этим основанием (например, аммиачный буфер
), или слабое основание и соль, образованную сильной кислотой и этим основанием (например, аммиачный буфер 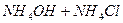 ). Эти растворы обладают определенными свойствами, которые проиллюстрируем на примере ацетатного буфера.
). Эти растворы обладают определенными свойствами, которые проиллюстрируем на примере ацетатного буфера.
Присутствие ацетата натрия (сильного электролита), который полностью диссоциирован, настолько увеличивает концентрацию ионов CH  COO
COO  , что, в соответствии с принципом Ле-Шателье, диссоциация уксусной кислоты полностью подавляется:
, что, в соответствии с принципом Ле-Шателье, диссоциация уксусной кислоты полностью подавляется:
В результате можно считать, что в буферном растворе активность анионов равна активности анионов соли 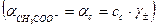 , а активность кислоты равна ее концентрации
, а активность кислоты равна ее концентрации 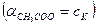 . Подставляя эти величины в выражение константы диссоциации, логарифмируя и вводя обозначение
. Подставляя эти величины в выражение константы диссоциации, логарифмируя и вводя обозначение 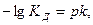 получимследующие формулы:
получимследующие формулы:
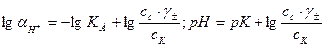 (32.9)
(32.9)
Эти формулы показывают, что pH буферного раствора зависит от константы диссоциации кислоты и соотношения аналитических концентраций соли и кислоты. При разбавлении буферного раствора это соотношение не меняется, а незначительное повышение pH обусловлено изменением коэффициента активности соли. Добавление сильной кислоты тоже сравнительно слабо отражается на изменении pH. При добавлении сильной кислоты к буферному раствору идет реакция с образованием недиссоциированной уксусной кислоты:
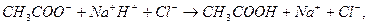 а при добавлении сильного основания- реакция нейтрализации:
а при добавлении сильного основания- реакция нейтрализации:
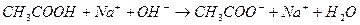 .
.
Ионы 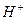 в первом случае, и ионы
в первом случае, и ионы 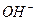 во втором, связываются в малодиссоциированные молекулы (
во втором, связываются в малодиссоциированные молекулы ( 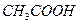 и
и 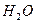 ), в результате чего в pH раствора практически не изменяется.
), в результате чего в pH раствора практически не изменяется.
Способность буферных растворов противостоять изменению pH количественно выражается величиной, называемой буферной емкостью. Буферная емкость — это количество кислоты или щелочи которое нужно добавить к раствору, чтобы изменить его pH на единицу.
Числа переноса
Каждый вид ионов переносит определенное количество электричества, зависящее от заряда и концентрации ионов, а также скорости их движения в электрическом поле. Отношение количества электричества 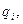 перенесенного ионами
перенесенного ионами 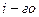 вида, к общему количеству электричества
вида, к общему количеству электричества 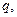 перенесенному всеми ионами, находящимися в растворе, называют числом переноса ионов:
перенесенному всеми ионами, находящимися в растворе, называют числом переноса ионов:
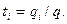 (32.42)
(32.42)
В соответствии с этим определением сумма чисел переноса всех видов ионов в растворе равна единице.
Для симметричного электролита KA, диссоциирующего на два вида ионов 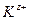 и
и  , количество электричества, перенесенное катионами и анионами, составит соответственно:
, количество электричества, перенесенное катионами и анионами, составит соответственно:
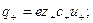
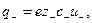 (32.43)
(32.43)
где 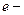 элементарный заряд;
элементарный заряд; 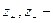 заряд катиона и аниона;
заряд катиона и аниона;  молярная концентрация катионов и анионов
молярная концентрация катионов и анионов 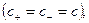
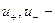 абсолютные скорости ионов. Отношение чисел переноса катионов
абсолютные скорости ионов. Отношение чисел переноса катионов  и анионов
и анионов 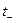 равно отношению их абсолютных скоростей или подвижностей:
равно отношению их абсолютных скоростей или подвижностей:
 ,
,
а поскольку 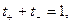 то
то
 и
и 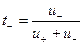 (32.44)
(32.44)
Из уравнений видно, что число переноса данного вида иона зависит от абсолютной скорости и подвижности обоих видов ионов, т. е. в растворах разных электролитах числа переноса одного и того же иона различны.
На степень гидратации ионов, величины их абсолютной скорости и числа переноса влияют концентрация раствора и температура. С ростом концентрации примерно до 0,1 моль/л для большинства электролитов числа переноса ионов изменяются незначительно; в области более высоких концентраций это изменение заметнее. При повышении температуры размеры гидратных оболочек слабо гидратированных ионов уменьшаются менее резко, чем сильно гидрати-рованных (а иногда даже увеличиваются). В результате величины абсолютной подвижности катионов и анионов сближаются, и их числа переноса стремятся к 0,5.
[1] Диэлектрическая проницаемость — величина, показывающая, во сколько раз сила взаимодействия двух зарядов в изучаемой среде меньше, чем в вакууме. [2] Зарядом иона z называют отношение заряда иона, выраженного в кулонах, к заряду электрона Кл; заряд иона, в кулонах, соответственно, равен произведению ez. [3] Далее во всех случаях, где это особо не оговаривается, с целью упрощения мы будем говорить о коэффициенте активности и активности электролитов, понимая, что речь идет о среднем коэффициенте активности и средней активности. В дальнейшем пренебрегается и различием между тремя способами выражения активности (коэффициента активности), что вполне допустимо для разбавленных растворов. [4] Используют также определение – радиус (толщина) ионной атмосферы, дебаевский радиус. [5] Обозначение единицы электрической проводимости сименс, как и всех других единиц, происходящих от имен собственных, пишется с прописной буквы (См). Это обозначение нельзя путать с обозначением единицы измерения длины – сантиметр (см).Физическая и химическая теории растворов.
Растворы являются сложными системами, в которых имеют место различные виды взаимодействия между частицами (Ван-дер-Ваальсовы, электростатические и т.д.).
Существуют две точки зрения на природу растворения и растворов. Согласно физической точке зрения, растворение является чисто физическим процессом (разрушение кристаллической решетки при растворении твердых тел). Растворы при этом рассматриваются как молекулярные смеси нескольких веществ, не взаимодействующих химически. Противоположные представления были развиты Д. И. Менделеевым, который считал растворение химическим процессом, а растворы рассматривал как непрочные соединения компонентов раствора, находящихся в состоянии частичной диссоциации и отличающихся от обычных соединений переменным составом.
В настоящее время используются представления обеих теорий и доминирующая роль физической или химической компонент, в процессе растворения, определяется свойствами растворителя и растворенного вещества (системы).


Папиллярные узоры пальцев рук — маркер спортивных способностей: дерматоглифические признаки формируются на 3-5 месяце беременности, не изменяются в течение жизни.

Механическое удерживание земляных масс: Механическое удерживание земляных масс на склоне обеспечивают контрфорсными сооружениями различных конструкций.

Общие условия выбора системы дренажа: Система дренажа выбирается в зависимости от характера защищаемого.
Физическая теория растворов.
РАСТВОРЫ
Общие сведения
Растворы — это гомогенные системы переменного состава, состоящие из двух и более веществ, называемых компонентами. По агрегатному состоянию растворы могут быть газообразными (воздух), жидкими (кровь, лимфа) и твердыми (сплавы). В медицине наибольшее значение имеют жидкие растворы, которые играют исключительную роль в жизнедеятельности живых организмов. С образованием растворов связаны процессы усвоения пищи и выведения из организма продуктов жизнедеятельности. В форме растворов вводится большое количество лекарственных препаратов.
Для качественного и количественного описания жидких растворов используются термины «растворитель» и «растворенное вещество», хотя в некоторых случаях такое разделение является достаточно условным. Так, медицинский спирт (96% раствор этанола в воде) скорее следует рассматривать как раствор воды в спирте. Все растворители делятся на неорганические и органические. Важнейшим неорганическим растворителем (а в случае биологических систем – единственным) является вода. Это обусловлено такими свойствами воды, как полярность, низкая вязкость, склонность молекул к ассоциации, относительно высокие температуры кипения и плавления. Растворители органической природы разделяют на полярные (спирты, альдегиды, кетоны, кислоты) и неполярные (гексан, бензол, четыреххлористый углерод).
Процесс растворения в равной степени зависит как от природы растворителя, так и от свойств растворенного вещества. Очевидно, что способность образовывать растворы выражена у разных веществ по-разному. Одни вещества могут смешиваться друг с другом в любых количествах (вода и этанол), другие – в ограниченных (вода и фенол). Однако, следует помнить: абсолютно нерастворимых веществ не существует!
Склонность вещества растворяться в том или ином растворителе можно определить, используя простое эмпирическое правило: подобное растворяется в подобном. Действительно, вещества с ионным (соли, щелочи) или полярным (спирты, альдегиды) типом связи хорошо растворимы в полярных растворителях, например, в воде. И наоборот, растворимость кислорода в бензоле на порядок выше чем в воде, так как молекулы O2 и C6H6неполярны.
Степень сродства соединения к определенному типу растворителя можно оценить, анализируя природу и количественное соотношение входящих в его состав функциональных групп, среди которых выделяют гидрофильные (притягивающие воду) и гидрофобные (отталкивающие воду). К гидрофильным относят полярные группы, такие как гидроксильная (-OH), карбоксильная (-COOH), тиольная (-SH), амино (-NH2). Гидрофобными считают неполярные группы: углеводородные радикалы алифатического (-CH3, -C2H5) и ароматического (-C6H5) рядов. Соединения, имеющие в своем составе как гидрофильные, так и гидрофобные группы, называют дифильными. К таким соединениям относят аминокислоты, белки, нуклеиновые кислоты.
Теории растворов
В настоящее время известны две основные теории растворов: физическая и химическая.
Физическая теория растворов.
Физическая теория растворов была предложена С. Аррениусом (1883) и Я. Г. Вант-Гоффом (1885). В данной теории растворитель рассматривается как химически инертная среда, в которой равномерно распределены частицы (молекулы, ионы) растворенного вещества. При этом предполагается отсутствие межмолекулярного взаимодействия как между частицами растворенного вещества, так и между молекулами растворителя и частицами растворенного вещества. Однако впоследствии выяснилось, что условиям данной модели удовлетворяет поведение лишь малой группы растворов, которые были названы идеальными. В частности, идеальными растворами можно считать газовые смеси и очень сильно разбавленные растворы неэлектролитов.
Химическая теория растворов.
Химическая, или сольватная, теория растворов была предложена в 1887 г. Д.И. Менделеевым, который установил, что в реальном растворе присутствуют не только индивидуальные компоненты, но и продукты их взаимодействия. Исследования водных растворов серной кислоты и этилового спирта, проведенные Д.И. Менделеевым, легли в основу теории, суть которой заключается в том, что между частицами растворенного вещества и молекулами растворителя происходят взаимодействия, в результате которых образуются нестойкие соединения переменного состава, называемые сольватами или гидратами, если растворителем является вода. Главную роль в образовании сольватов играют непрочные межмолекулярные силы, в частности, водородная связь.
В этой связи следует принять следующую трактовку понятия «раствор»:
Раствором называется гомогенная система переменного состава, состоящая из двух и более компонентов и продуктов их взаимодействия.
Из данного определения следует, что растворы занимают промежуточное положение между химическими соединениями и смесями. С одной стороны, растворы однородны, что позволяет рассматривать их как химические соединения. С другой стороны, в растворах нет строгого стехиометрического соотношения между компонентами. Кроме того, растворы можно разделить на составные части (например, при упаривании раствора NaCl можно выделить соль в индивидуальном виде).
Связь между различными способами
Кислоты и основания
Несмотря на то, что понятия «кислота» и «основание» широко используются для описания химических процессов, единого подхода к классификации веществ с точки зрения отнесения их к кислотам или основаниям нет. Существующие в настоящее время теории (ионная теория С. Аррениуса, протолитическая теория И. Бренстеда и Т. Лоури и электронная теория Г. Льюиса) имеют определенные ограничения и, таким образом, применимы лишь в частных случаях. Остановимся подробнее на каждой из этих теорий.
Теория Аррениуса.
В ионной теории Аррениуса понятия «кислота» и «основание» тесно связаны с процессом электролитической диссоциации:
Кислотой является электролит, диссоциирующий в растворах с образованием ионов Н + ;
Основаниемявляется электролит, диссоциирующий в растворах с образованием ионовОН — ;
Амфолитом (амфотерным электролитом) является электролит, диссоциирующий в растворах с образованием как ионовН + , так и ионов ОН — .
|
НА ⇄ Н + + А —
|
МеОН⇄Ме + + ОН —
|
nH + +MeOn n — ⇄Ме(ОН)n⇄Ме n + +nОН —
В соответствии с ионной теорией кислотами могут быть как нейтральные молекулы, так и ионы, например:
Аналогичные примеры можно привести и для оснований:
КОН К + + ОН —
К амфолитам относят гидроксиды цинка, алюминия, хрома и некоторые другие, а также аминокислоты, белки, нуклеиновые кислоты.
В целом, кислотно-основное взаимодействие в растворе сводится к реакции нейтрализации:
H + + ОН — H2O
Однако, ряд экспериментальных данных показывает ограниченность ионной теории. Так, аммиак, органические амины, оксиды металлов типа Na2O, СаО, анионы слабых кислот и т.д. в отсутствии воды проявляют свойства типичных оснований, хотя не имеют в своем составе гидроксид-ионов.
С другой стороны, многие оксиды (SO2, SO3, Р2О5 и т.д.), галогениды, галогенангидриды кислот, не имея в своем составе ионов водорода, даже в отсутствии воды проявляют кислотные свойства, т.е. нейтрализуют основания.
Кроме того, поведение электролита в водном растворе и в неводной среде может быть противоположным.
Так, CH3COOH в воде является слабой кислотой:
а в жидком фтороводороде проявляет свойства основания:
Исследования подобных типов реакций и в особенности реакций, протекающих в неводных растворителях, привели к созданию более общих теорий кислот и оснований.
Теория Бренстеда и Лоури.
Дальнейшим развитием теории кислот и оснований явилась предложенная И. Бренстедом и Т. Лоурипротолитическая (протонная) теория. В соответствии с этой теорией:
Кислотой называют любое вещество, молекулы (или ионы) которого способны отдавать протон, т.е. быть донором протона;
Основанием называют любое вещество, молекулы (или ионы) которого способны присоединять протон, т.е. быть акцептором протона;
Таким образом, понятие основания значительно расширяется, что подтверждается следующими реакциями:
ОН — + Н + Н2О
NH3+H + NH4 +
H2N-NH3 + +H + H3N + -NH3 +
По теории И. Бренстеда и Т. Лоури кислота и основание составляют сопряженную пару и связаны равновесием:
КИСЛОТА ⇄ ПРОТОН + ОСНОВАНИЕ
Поскольку реакция переноса протона (протолитическая реакция) обратима, причем в обратном процессе тоже передается протон, то продукты реакции являются друг по отношению к другу кислотой и основанием. Это можно записать в виде равновесного процесса:
где НА – кислота, В – основание, ВН + – кислота, сопряженная с основанием В, А — – основание, сопряженное с кислотой НА.
Примеры.
HCl и H2O – кислоты, Cl — и OH — – соответствующие сопряженные с ними основания;
HSO4 — и H3O + – кислоты, SO4 2 — и H2O – основания;
NH4 + – кислота, NH2 — – основание, а NH3 выступает в роли как кислоты (одна молекула), так и основания (другая молекула), т.е. демонстрирует признаки амфотерности – способности проявлять свойства кислоты и основания.
Такой способностью обладает и вода:
Здесь одна молекула Н2О присоединяет протон (основание), образуя сопряженную кислоту – ион гидроксония Н3О + , другая отдает протон (кислота), образуя сопряженное основание ОН — . Этот процесс называется автопротолизом.
Из приведенных примеров видно, что в отличие от представлений Аррениуса, в теории Бренстеда и Лоури реакции кислот с основаниями не приводят к взаимной нейтрализации, а сопровождаются образованием новых кислот и оснований.
Необходимо также отметить, что протолитическая теория рассматривает понятия «кислота» и «основание» не как свойство, но как функцию, которую выполняет рассматриваемое соединение в протолитической реакции. Одно и то же соединение может в одних условиях реагировать как кислота, в других – как основание. Так, в водном растворе СН3СООН проявляет свойства кислоты, а в 100%-й H2SO4 – основания.
Однако, несмотря на свои достоинства, протолитическая теория, как и теория Аррениуса, не применима к веществам, не содержащим атомов водорода, но, в тоже время, проявляющим функцию кислоты: галогенидам бора, алюминия, кремния, олова.
Теория Льюиса.
Иным подходом к классификации веществ с точки зрения отнесения их к кислотам и основаниям явилась электронная теория Льюиса. В рамках электронной теории:
кислотой называют частицу (молекулу или ион), способную присоединять электронную пару (акцептор электронов);
основанием называют частицу (молекулу или ион), способную отдавать электронную пару (донор электронов).
Согласно представлениям Льюиса, кислота и основание взаимодействуют друг с другом с образованием донорно-акцепторной связи. В результате присоединения пары электронов у атома с электронным дефицитом возникает завершенная электронная конфигурация — октет электронов. Например:
Аналогичным образом можно представить и реакцию между нейтральными молекулами:
Реакция нейтрализации в терминах теории Льюиса рассматривается как присоединение электронной пары гидроксид-иона к иону водорода, предоставляющему для размещения этой пары свободную орбиталь:
Таким образом, сам протон, легко присоединяющий электронную пару, с точки зрения теории Льюиса, выполняет функцию кислоты. В этой связи, кислоты по Бренстеду могут рассматриваться как продукты реакции между льюисовскими кислотами и основаниями. Так, HCl является продуктом нейтрализации кислоты H + основанием Cl — , а ион H3O + образуется в результате нейтрализации кислоты H + основанием H2O.
Реакции между кислотами и основаниями Льюиса также иллюстрируют следующие примеры:
| Кислота | Основание | Кислотно-основный комплекс |
| SO2 | H2O | H2SO3 |
| Al(OH)3 | OH – | [Al(OH)4] – |
| Zn(OH)2 | 2OH – | [Zn(OH)4] 2 – |
| Ag + | 2CN – | [Ag(CN)2] – |
К основаниям Льюиса также относят галогенид-ионы, аммиак, алифатические и ароматические амины, кислородсодержащие органические соединения типа R2CO, (где R- органический радикал).
К кислотам Льюиса относят галогениды бора, алюминия, кремния, олова и других элементов.
Очевидно, что в теории Льюиса понятие «кислота» включает в себя более широкий круг химических соединений. Это объясняется тем, что по Льюису отнесение вещества к классу кислот обусловлено исключительно строением его молекулы, определяющим электронно-акцепторные свойства, и не обязательно связано с наличием атомов водорода. Льюисовские кислоты, не содержащие атомов водорода, называют апротонными.
РАСТВОРЫ
Общие сведения
Растворы — это гомогенные системы переменного состава, состоящие из двух и более веществ, называемых компонентами. По агрегатному состоянию растворы могут быть газообразными (воздух), жидкими (кровь, лимфа) и твердыми (сплавы). В медицине наибольшее значение имеют жидкие растворы, которые играют исключительную роль в жизнедеятельности живых организмов. С образованием растворов связаны процессы усвоения пищи и выведения из организма продуктов жизнедеятельности. В форме растворов вводится большое количество лекарственных препаратов.
Для качественного и количественного описания жидких растворов используются термины «растворитель» и «растворенное вещество», хотя в некоторых случаях такое разделение является достаточно условным. Так, медицинский спирт (96% раствор этанола в воде) скорее следует рассматривать как раствор воды в спирте. Все растворители делятся на неорганические и органические. Важнейшим неорганическим растворителем (а в случае биологических систем – единственным) является вода. Это обусловлено такими свойствами воды, как полярность, низкая вязкость, склонность молекул к ассоциации, относительно высокие температуры кипения и плавления. Растворители органической природы разделяют на полярные (спирты, альдегиды, кетоны, кислоты) и неполярные (гексан, бензол, четыреххлористый углерод).
Процесс растворения в равной степени зависит как от природы растворителя, так и от свойств растворенного вещества. Очевидно, что способность образовывать растворы выражена у разных веществ по-разному. Одни вещества могут смешиваться друг с другом в любых количествах (вода и этанол), другие – в ограниченных (вода и фенол). Однако, следует помнить: абсолютно нерастворимых веществ не существует!
Склонность вещества растворяться в том или ином растворителе можно определить, используя простое эмпирическое правило: подобное растворяется в подобном. Действительно, вещества с ионным (соли, щелочи) или полярным (спирты, альдегиды) типом связи хорошо растворимы в полярных растворителях, например, в воде. И наоборот, растворимость кислорода в бензоле на порядок выше чем в воде, так как молекулы O2 и C6H6неполярны.
Степень сродства соединения к определенному типу растворителя можно оценить, анализируя природу и количественное соотношение входящих в его состав функциональных групп, среди которых выделяют гидрофильные (притягивающие воду) и гидрофобные (отталкивающие воду). К гидрофильным относят полярные группы, такие как гидроксильная (-OH), карбоксильная (-COOH), тиольная (-SH), амино (-NH2). Гидрофобными считают неполярные группы: углеводородные радикалы алифатического (-CH3, -C2H5) и ароматического (-C6H5) рядов. Соединения, имеющие в своем составе как гидрофильные, так и гидрофобные группы, называют дифильными. К таким соединениям относят аминокислоты, белки, нуклеиновые кислоты.
Теории растворов
В настоящее время известны две основные теории растворов: физическая и химическая.
Физическая теория растворов.
Физическая теория растворов была предложена С. Аррениусом (1883) и Я. Г. Вант-Гоффом (1885). В данной теории растворитель рассматривается как химически инертная среда, в которой равномерно распределены частицы (молекулы, ионы) растворенного вещества. При этом предполагается отсутствие межмолекулярного взаимодействия как между частицами растворенного вещества, так и между молекулами растворителя и частицами растворенного вещества. Однако впоследствии выяснилось, что условиям данной модели удовлетворяет поведение лишь малой группы растворов, которые были названы идеальными. В частности, идеальными растворами можно считать газовые смеси и очень сильно разбавленные растворы неэлектролитов.
Теории растворов.
Существует 2 теории растворов: физическая и химическая.
Физическая теория растворов.
Была открыта Якобом Г. Вант-Гоффом и Свате А.Аррениусом.
Суть теории растворов: растворитель – химическая индифферентная среда, в которой равномерно распределены частицы растворенного вещества. Теория не предполагает наличие межмолекулярных связей между растворителем и растворенным веществом.
Под эту теорию подходят только идеальные растворы, где компоненты растворителя никак не воздействуют на растворимое соединение. Примером являются газовые растворы, где нереагирующие между собой газы смешиваются друг с другом в неограниченных количествах. Все физические данные (температура кипения и плавления, давление, теплоемкость) вычисляется исходя из свойств всех соединений, входящих в состав.
По закону Дальтона: общее давление газовой смеси равно сумме парциальных давлений ее компонентов:
Химическая теория растворов.
Химическую (сольватную) теорию растворов описал Д.И. Менделеев. Суть заключается в следующем: частицы растворителя и растворенного вещества реагируют друг с другом, в результате чего получаются нестойкие соединения переменного состава – гидраты (сольваты). Основные связи тут – водородные.
Вещество может распадаться на слои (растворяться) в случае полярного растворителя (воды). Ярким примером является растворение поваренной соли.
Также может проткать реакция между компонентами смеси:
В ходе процесса растворения происходит изменение состава и объема реакционной смеси, т.к. протекают 2 процесса: разрушение структуры растворяемого вещества и химическая реакция между частицами. Оба процесса идут с изменением энергии.
Тепловые эффекты могут быть экзотермическими и эндотермическими (с выделением и поглощением энергии).
Соединения с частицами растворителя называются гидратами.
Кристаллические вещества, в состав которых входят гидраты, называются кристаллогидратами и имеют различную окраску. Например, кристаллогидрат сульфата меди: CuSO4 ·5H2О. Раствор кристаллогидрата синий. Если рассмотреть кристаллогидрат кобальта CoCl2· 6H2O, то он обладает розовым цветом, CoCl2· 4H2O – красный, CoCl2 ·2H2O — сине-фиолетовый, CoCl2 ·H2О – темно-синий, а безводный раствор хлорида кобальта – бледно-синего цвета.
Растворы — системы, состоящие из молекул, атомов и(или) ионов неск. разл. типов, при этом числа разл. частиц не находятся в к—л. определённых стехиометрич. соотношениях друг с другом (что отделяет растворы от хим. соединений). К растворам обычно относят такие многокомпонентные системы, в к-рых при неизменных внеш. условиях достигается состояние термодинамич. равновесия.
Агрегатное состояние растворов может быть твёрдым (твёрдые растворы), жидкокристаллическим (жидкие кристаллы ),жидким или газообразным. Будучи макроскопически пространственно однородными, на молекулярных масштабах растворы могут обладать своеобразной микроструктурой (микрогетерогенные растворы, или ассоциирующие коллоиды), к-рая определяется температурой, давлением и составом раствора. Если микроструктура раствора является регулярной (в одном, двух или трёх измерениях), то его относят к лиотропным жидким кристаллам. Жидкие растворы с нерегулярной микроструктурой (обычно многокомпонентные, содержащие органич. вещества и соли) наз. эмульсиями (микроэмульсиями). Суспензии частиц размером от неск. нм до тысяч нм относят к коллоидным растворам.
В том случае, когда молекулы растворённого вещества диссоциируют на ионы, растворы относят к особому классу — растворы электролитов. Отличит. свойствами обладают растворы полимеров.
Термодинамические свойства растворов
Термодинамич. свойства растворов описываются общими для многокомпонентных систем соотношениями термодинамики. Число веществ n, кол-ва к-рых в состоянии полного термодинамич. равновесия могут быть заданы произвольно, наз. числом независимых компонент раствора. Если число молекул (атомов) одной из компонент системы N намного превышает числа N1, . Nn-1 молекул остальных компонент, раствор наз. разбавленным (слабым). Вещество, содержащее N частиц, в этом случае наз. растворителем, остальные компоненты — растворёнными веществами. Величины
ci = Ni/N (здесь ) наз. молярными (молекулярными) концентрациями (используются также весовые и объёмные концентрации). Согласно Гиббса правилу фаз, в системе, состоящей из n компонент, в равновесии не может находиться более n + 2 фаз. Состояние раствора описывается n + 1 переменной (n -1 значение концентраций, темп-pa Т и давление р)и может быть изображено точкой в n + 1-мерном пространстве. Если в этом пространстве построить гиперповерхности меньшего числа измерений, на к-рых выполняются условия равновесия двух или большего числа фаз, то получится поверхность, характеризующая состояние системы,- т. н. фазовая диаграмма системы (или диаграмма состояния ).Обычно пользуются сечениями фазовой диаграммы различными плоскостями.
Диаграммы плавления и кипения растворов. В отличие от чистых веществ, изменение агрегатного состояния раствора происходит в нек-ром интервале изменения концентраций компонент, температуры и(или) давления. Простейший случай равновесия двух фаз реализуется, когда обе компоненты, образующие раствор, в обеих фазах смешиваются в произвольных отношениях. Кривые равновесия в этом случае не имеют максимумов и минимумов и образуют характерную «сигару» (диаграмма Т — с, с — концентрация; рис. 1). Пусть для определённости рассматриваемые фазы представляют собой жидкость (низкотемпературная фаза II) и пар (высокотемпературная фаза I). Если изображающая точка системы (Т, с)лежит выше кривой FAG, то агрегатное состояние системы — пар, если ниже кривой FCG — жидкость. Заштрихованная область между кривыми FAG и FCG соответствует равновесию двух фаз (представляющих собой т. н. насыщенные растворы),
концентрации к-рых характеризуются растворимостью веществ и равны с’ и с», в точке В массы определяются «правилом рычага», согласно к-рому кол-ва молекул в фазах I и II обратно пропорциональны длине отрезков соответственно АВ и ВС:
В случае равновесия системы жидкость — пар кривая FAG наз. кривой конденсации, a FCG — кривой кипения. В случае равновесия твёрдой и жидкой фаз кривая FAG наз. кривой ликвидуса, a FCG — кривой солидуса.
Фазовые диаграммы типа «сигары» дают растворы веществ, близких по химическим свойствам: для диаграмм плавления — это, например, бинарные (двухкомпонентные) растворы Ge — Si, Ag — Аи, Си — Ni, AgCl — NaCl, для диаграмм кипения — системы бензол — толуол, этиловый спирт — вода и другие. При относительно небольшом хим. отличии смешиваемых веществ А а В кривые кипения и конденсации могут иметь максимум или минимум, в к-ром эти кривые касаются друг друга (рис. 2). Раствор, состав к-рого соответствует точке касания (точка А), наз. азеотропным. Фазовый переход (кипение или плавление) в растворе такого состава происходит так же, как в чистом веществе,- целиком. Когда точка касания является максимумом кривых равновесия (рис. 2, о), кипение раствора произвольного нач. состава приводит к смещению изображающей точки системы в положение А. Т. о., азеотропная точка является устойчивой предельной точкой процесса кипения. Если же касание кривых равновесия происходит в их минимуме (рис. 2,б), то в процессе фазового перехода при повышении температуры изображающая точка системы сдвигается к одному из чистых веществ; азеотропная точка достигается при охлаждении смеси.
Рис. 2. Примеры диаграмм состояния: а — теплота смешения в жидкости меньше, чем в паре; б — теплота смешения в твёрдой фазе больше, чем в жидкой.
При повышении p в системе жидкость — пар форма «сигары» изменяется, а при давлениях выше критического (см. Критическая точка)для одной из компонент, когда отсутствует различие между двумя фазами этого вещества, «сигара» вырождается в петлю путём смыкания кривых кипения и конденсации в нек-рой (критической) точке (рис. 3).
Рис. 3. Пример диаграммы состояния двухкомпонентной системы жидкость — дар в случае, когда давление в системе превышает критическое давление компоненты А. Разделение смеси на жидкую и газообразную фазы имеет смысл лишь в пределах заштрихованной области. К — критическая точка.
Диаграммы смешения растворов. Наряду с равновесиями фаз, находящихся в различных агрегатных состояниях, в растворе могут сосуществовать фазы, находящиеся в одном агрегатном состоянии, например жидком. На рис. 4 изображена диаграмма, соответствующая случаю ограниченной смешиваемости двух веществ в одной жидкой фазе. Жидкости полностью смешиваются в области, лежащей над кривой AKB, и ограниченно смешиваются в области, лежащей под этой кривой (где имеет место расслоение растворов на две жидкие фазы с составом, определяемым «правилом рычага»). Точка К максимума кривой — критическая: в окрестности этой точки наблюдаются аномалия теплоёмкости, критич. опалесценция и др. критические явления .Существуют жидкие системы (напр., триэтиламин — вода), для к-рых область неограниченной смешиваемости лежит ниже нек-рой кривой и к-рые имеют нижнюю температуру смешения, а также системы, имеющие как верхнюю, так и нижнюю температуры смешения (рис. 5).
Рис. 4. Диаграмма смешения системы фенол — вода. К — критическая точка смешения с параметрами Тк = 66 С, ск = 0,65.
Верхняя критич. темп-pa смешения растёт с увеличением различий хим. свойств смешиваемых веществ.
Рис. 5. Диаграмма смешения системы никотин — вода. Жидкости полностью смешиваются вне области, ограниченной замкнутой кривой.
Качественно эта зависимость выражается в понятии миксотропного ряда, в к-ром все вещества располагаются в соответствии со значением приписываемого каждому из них т. н. эффективного заряда е (табл.; численные значения е получены не для всех веществ, но они расположены в порядке убывания е). Верхняя темп-pa смешения растёт с увеличением разности «зарядов» смешиваемых веществ.
Миксотропный ряд
Эффективный заряд е, в относит. ед.
Если верхняя критич. темп-pa смешения веществ в низкотемпературной фазе оказывается выше температуры минимума кривых равновесия, изображённых на рис. 2 (б), то фазовая диаграмма системы приобретает вид, аналогичный изображённому на рис. 6. Такая диаграмма принадлежит к эвтектическому типу; раствор, имеющий состав, отвечающий точке эвтектики Е, наз. эвтектикой. Плавление эвтектики происходит при определ. температуре, как и плавление химически чистого вещества. В растворе с ограниченной взаимной растворимостью могут возникать также фазовые диаграммы перитектического типа (рис. 7). Пери-тектическая точка имеет самую высокую температуру плавления определённой твёрдой фазы, в то время как точка эвтектики — наинизшую температуру затвердевания определённой жидкой фазы. Эвтектическая и перитектическая точки — это точки сосуществования трёх фаз: жидкой и двух твёрдых.
Рис. 6. Фазовая диаграмма системы Sn — Pb эвтектического типа, a, b — твёрдые растворы. Точка пересечения трёх линий, в которых сосуществуют жидкая и две твёрдые фазы,- точка эвтектики.
Рис. 7. Фазовая диаграмма перитектического типа. P- перитектическая точка, a,b- твёрдые растворы.
Законы слабых растворов. Термодинамический потенциал слабого раствора имеет вид
где m(p, T)- химический потенциал чистого растворителя, yi(p, T) — нек-рая функция, зависящая от природы растворителя. Растворённое вещество распределяется между разл. фазами т. о., что отношение концентрации cI и сII в этих фазах зависит лишь от р, Т, но не от полного кол-ва растворённого вещества (т. н. закон распределения):
В частности, когда одна из фаз представляет собой газ, имеет место Генри закон ,согласно к-рому концентрация слабого раствора пропорциональна давлению газа р. При растворении в жидкости нелетучего вещества давление p насыщенного пара над раствором меньше, чем давление над чистым растворителем p:
(Рауля закон), где и — числа молей растворённого вещества и растворителя в единице объёма раствора. По отношению к нелетучему веществу поверхность жидкости ведёт себя как непроницаемая перегородка, и (1) представляет собой частный случай выражения для осмотического давления (см. Осмос)слабых растворов. При этом между частями системы, разделёнными перегородкой, проницаемой для растворителя, но непроницаемой для растворённых веществ, возникает разность давлений
где V — объём части сосуда, занятой растворёнными веществами.
Из закона Рауля (1) следует, что при пост. давлении темп-pa кипения раствора выше температуры кипения чистого растворителя :
где m — молекулярная масса веществ растворителя, qLV — уд. теплота испарения. Темп-pa замерзания раствора ниже температуры замерзания ТЗ чистого растворителя:
Ассоциирующие растворы
Мицеллообразование. Физико-химического свойства раствора широкого класса веществ, молекулы к-рых имеют асиммет-рич. форму,- т. н. амфифильные вещества (наз. также дифильными или поверхностно-активными веществами), определяются образованием в них т. н. мицелл — агрегатов молекул растворённого вещества. Такие растворы называются мицеллярными.
Амфифильные вещества имеют вытянутые молекулы (часто линейные) длиной 20-30 , имеющие хорошо выраженные гидрофильные пли олеофильные (жирные, гидрофобные) части (см. Гидрофильность и гидрофоб-ность)К таким веществам относятся соли жирных к-т (напр., мыло -стеарат натрия), имеющие в составе молекулы гибкую парафиновую цепь СnН2n+1 («жирный хвост»), присоединённую к полярной группе — «головке» «Головка» образована группой атомов, соединенных полярными связями. Амфифильными молекулами являются также липиды и фосфолипиды, входящие в состав клеточных мембран (си. Клеточные структуры).
Участок диаграммы состояния раствора амфифильного вещества приведён на рис. 8. Кривая равновесия АВС отделяет области жидких фаз I (молекулярный раствор) и II (мицеллярный раствор, или ассоциирующий коллоид, см. ниже) от твёрдой фазы III. Области молекулярного и мицеллярного раствора разделены нек-рой переходной областью BD, а не линией, как в обычном случае равновесия двух фаз. Мин. темп-pa , при к-рой возможно образование мицелл, наз. температурой Крафта. Концентрация амфифильного вещества с*, при к-рой начинается образование мицелл, наз. критической концентрацией мицел-лообразования (ККМ). ККМ сильно зависит от температуры, ионной силы раствора (сумма значений концентраций всех ионов раствора, умноженных на квадрат их зарядов) и т. д. ККМ падает с ростом длины «жирного хвоста» молекулы приближённо линейно при числе атомов углеводородной цепи n -3 см -3
Форма и структура мицелл зависят от типа растворителя (полярный или гидрофобный), от концентрации P его состава, температуры и др. Вблизи ККМ форма мицелл близка к сферической. В полярной среде (напр., в воде) мицеллы имеют углеводородное ядро, а полярные «головки» обращены к молекулам воды. В жирной среде мицелла имеет обращённую структуру: полярные «головки» образуют ядро, а олеофильные «хвосты» контактируют с растворителем (рис. 10). Каждая мицелла в Р. содержит 20-100 молекул амфифильного вещества (т.н. число агрегации). Обычно часть полярных «головок» молекул диссоциирована на ионы (для ионногенных амфифильных веществ).
Рис 10. Структура агрегатов в растворах амфифильного веще-ства· а — гибсовский монослой на поверхности раздела жирной и полярной сред; б — сферическая мицелла в полярном растворителе; в — обращённая сферическая мицелла в жирной среде; г — ламелла (бислой) в полярной среде; д — обращенная ла-мелла (мыльная плёнка); е — пузырёк (везикула), образованный в полярной среде.
С ростом концентрации Р. форма мицелл может изменяться: они принимают цилиндрическую, дискообразную форму или форму трёхосного эллипсоида. Это проявляется в изменении индикатрисы рассеяния света Р. и в изменении зависимости вязкости Р. от концентрации. В нек-рых случаях в Р. могут присутствовать ци-линдрич. мицеллы, содержащие
10 3 -10 4 молекул. В области концентраций Р., превышающих ККМ, именно мицеллы являются элементарными структурными единицами, определяющими физ. свойства возникающих в Р. фаз, как изотропных, так и жидкокристаллических (см. ниже).
Надмолекулярные жидкокристаллические структуры в растворах. Подобно молекулярным, мицеллярные Р. при нек-рой концентрации мицелл могут расслаиваться. Вблизи критич. точек расслоения (к-рые могут быть как верхними, так и нижними) наблюдаются критич. явления. Отслаивающаяся при увеличении концентрации более плотная фаза может быть как изотропной, так и анизотропной (см. Жидкие кристаллы). В бинарных системах обычно возникают гексагональная, ламеллярная (смектическая) и (или) кубическая фазы. Переход между ними происходит вследствие изменения формы или (и) размеров мицелл.
Гексагональные фазы образованы цилиндрич. мицеллами неопределённой длины, упакованными в двумерную решётку, имеющую гексагональную симметрию (рис. 11). Структура мицелл может быть нормальной или обращённой в зависимости от типа амфифильного вещества и растворителя. Радиус нормальных цилиндрич. мицелл примерно на 10- 30% меньше длины
полностью вытянутой амфифильной молекулы, расстояние между цилиндрами в зависимости от содержания воды изменяется в пределах 8-50. Для обращённых мицелл диаметр водного цилиндра составляет 10-20 , расстояние между ними ок. полутора длин «жирных хвостов» амфифильных молекул.
Ламеллярные (смектические)фазы образованы дискообразными мицеллами неопределённого диаметра (ламсллами). Толщина ламеллы на 10- 30% меньше удвоенной длины амфифильной молекулы (ряс. 12), величина водного промежутка между ламеллами изменяется при их набухании. Если данное вещество образует в Р. гексагональные фазы с мицеллами как нормального, так и обращённого типов, то ламеллярная фаза располагается на фазовой диаграмме в области концентраций, промежуточных между двумя гексагональными фазами.
В зависимости от температуры и состава Р. амфи-фильные молекулы в ла-медлах могут находиться в «расплавленном» или «кристаллическом» состоянии. Фаза ламеллярного типа, возникающая при затвердевании парафиновых цепей молекул при понижении температуры, носит название фазы геля. Толщина ламеллы в этих фазах может составлять одну или две длины полностью вытянутой амфифильной молекулы. Молекулы упакованы в двумерную гексагональную решётку; площадь, приходящаяся на одну молекулу в бислое, близка к минимально возможной (при одной парафиновой цепи в «жирном хвосте» — ок. 20). Длинные оси молекул могут быть наклонены по отношению к нормали амфифильного слоя. Водный промежуток составляет обычно 10-20, но может возрастать до «200(т. н. неограниченное набухание), если в системе присутствуют ионогенные амфифильные молекулы, подобные молекулам жирных кислот. Т. о., согласно классификации, принятой для термотропных жидких кристаллов, гели соответствуют смектич. фазам типов ВA и BC.
При добавлении в систему воды ламеллярные фазы втягивают воду — набухают. При этом возможны два типа набухания. В первом случае весь добавленный растворитель проникает в пространство между поляр-выми «головками» амфифильных молекул, что приводит к увеличению уд. площади, приходящейся на одну молекулу в ламелле. Период ламеллярной структуры остаётся примерно постоянным. Во втором случае при набухании происходит увеличение периода структуры, пропорциональное кол-ву добавленной воды, при пост. уд. площади на молекулу. Возможны также промежуточные типы набухания лиотропных смектич. фаз. Тип набухания не зависит от длины «жирного хвоста» молекулы в гомологич. ряду данного вещества, а определяется гл. обр. видом её полярной «головки» и «обменным» катионом (ионом щелочного металла, входящим в её состав). Напр., уд. площадь, приходящаяся на полярную «головку» молекулы мыла, при прочих равных условиях возрастает в ряду «обменных» катионов Na + , К + , Rb + , Cs + .
Кубические фазы могут возникать при концентрациях Р., промежуточных между концентрациями гексагональной фазы и изотропного мицеллярного Р. либо между концентрациями ламеллярной и гексагональной фаз (рис. 13). Кубич. фазы, обладающие трёхмерной периодичностью, по существу являются не жидкокристаллическими, а относятся к т. н. пластич. кристаллам. Вследствие больших коэф. вязкости (h ! 10 2 пуаз) н изотропии оптич. и магн. свойств ку-бич. фазы наз. также вязкими изотропными фазам и. Для фаз, расположенных между гексагональной фазой и мицеллярным Р., наиб. вероятна структура с кубич. плотной упаковкой сферич. мицелл (рис. 14), а для фаз, расположенных в области концентраций между гексагональной и ламеллярной фазами, предложены т. н. биконтинуальные структуры, для к-рых поверхность, образованная полярными «головками» амфифильных молекул, делит трёхмерное пространство на две взаимопроникающие области — полярную и гидрофобную. На рис. 15 изображена одна из возможных структур такого типа, поверхность полярных групп для к-рой представляет собой т. н. поверхность Шварца — поверхность, имеющую пост. отрицат. кривизну в каждой точке. Как правило, в бинарных системах определённое амфифильное вещество образует лишь нек-рые из перечисленных фаз. В многокомпонентных системах, содержащих кроме амфифильных и др. компоненты, возможно существование не только перечисленных, но и др. фаз (напр., нематических).
Рис. 13. Фазовая диаграмма растпора хлорида додециламмо-нпя и воде. I1 — кубическая фаза, образованная сферическими мицеллами; Н — гексагональная фаза; La — ламеллярная фаза; V1 — вязкая изотропная кубическая фаза.
Нематические фазы. Нематич. структуры (ориентацией-но упорядочен-ные; см. Дальний и ближний порядок)могут быть образованы мицеллами разл. типов: цилиндрическими, дискообразными или имеющими вид трёхосного эллипсоида. В зависимости от типа мицелл различают три типа нематич. структур, два из к-рых являются одноосными, а третий — двухосным. При изменении состава или (и) температуры Р. возможны переходы одного типа нематич. упорядочения в другие.
Тензоры диэлектрич. проницаемости eik и диамагн. восприимчивости для одноосных нематиков имеют два разл. собств. значения и (соответственно показатели преломления обыкновенной и необыкновенной эл—магн. волн также различаются, см. Анизотропия ).Во внеш. магн. поле ось нематиков, образованных цилиндрич. мицеллами, ориентируется параллельно полю, т. е. они имеют положит. анизотропию диамагн. проницаемости: Анизотропия показателя преломления для этих фаз отрицательна: Напротив, выделенная ось нематиков, образованных дисками, ориентируется перпендикулярно магн. полю; в этом случае c3 0. Знак диамагн. анизотропии определяется анизотропией магн. свойств углеводородных ядер мицелл: разностью восприимчивостей «жирных хвостов» вдоль и поперёк осп амфифильной молекулы и степенью упорядочения этих молекул относительно оси мицеллы. В частности, для веществ, молекулы к-рых содержат в составе «жирного хвоста» одно или неск. ароматич. колец, знак анизотропии магн. восприимчивости может быть противоположным указанному выше. Знак диэлектрич. проницаемости нематиков определяется величиной деполяризующего действия мицелл, находящихся в водной среде. По абс. величине значения анизотропии показателя преломления и диамагн. анизотропии, как правило, на 1-2 порядка меньше, чем вслучае термотропных нематиков, и составляют
При изменении направления внеш. магн. поля нема-тич. структуры переориентируются. Время соответствующего переходного процесса в магн. поле напряжённостью H
10 4 Э варьируется в пределах 1-10 4 с в зависимости от содержания воды в образце. Модули упругости лиотропных нематич. кристаллов на 1-2 порядка меньше, чем в случае термотропных жидких кристаллов: К = 10 -7 -10 -9 дн/см 2 . Их величина определяется энергией взаимодействия между мицеллами в Р., к-рая складывается из энергии ван-дер-ваальсовых межатомных взаимодействий и электростатич. энергии взаимодействия ионов, находящихся в Р. и на поверхности мицелл. В нематич. фазах расстояние между мицеллами (цилиндрами и дисками) составляет обычно неск. сотен А, и искажение структуры растворителя, вызванное присутствием мицелл, по-видимому, слабо сказывается на взаимодействии агрегатов. В нематич. структурах с длиной цилиндрич. мицелл
10 3 p более существенное значение имеет изгиб мицелл, энергия к-рого сравнима с энергией взаимодействия между мицеллами.
Дефекты и текстуры лиотропных жидких кристаллов. Ближний порядок, существующий в упаковке молекул в мицеллы, а также во взаимном расположении мицелл в Р., определяет особенности текстуры макроскопич. образца. Так, для нематич. лиотропных фаз осн. дефектами упаковки мицелл, к-рые определяют характерную картину изображения образца, получаемую с помощью оптич. поляризац. микроскопа, являются дисклинации .Структура дисклинации в лиотропных нематич. кристаллах такая же, как в термотропных.
В ламеллярных фазах наим. энергию имеют деформации структур, при к-рых величина водного промежутка постоянна и равна своему равновесному значению во всём объёме образца, за исключением особых линий. Этому условию удовлетворяют краевые дислокации и различные конфокальные домены, простейшими из к-рых являются миелиновые фигуры (многослойные структуры, представляющие собой слои, свёрнутые по спирали; рис. 16). Краевые дислокации, к-рых в лиотропных смектич. жидких кристаллах может быть, по крайней мере, два типа (рис. 17), приводят к образованию т. н. террас Гранжа-на, видимых в оптический и электронный микроскопы, а также к образованию разл. пучков и сеток.
Структура дефектов в ламеллярных фазах существенно зависит от содержания воды в образце н от температуры. Одноврем. с изменением текстуры происходит изменение электропроводности Р. и коэф. диффузии ионов. При охлаждении системы в момент образования геля вследствие наклона амфифильных молекул в биослоях может возникать волнообразная деформация ламелл (наблюдаемая в электронный микроскоп и проявляющаяся в особенностях рассеяния рентг. лучей).
Рис. 17. Два типа краевых дислокаций в лиотропных смектических структурах.
Микроэмульсии
Добавление в систему несмешиваемых в обычных условиях жидкостей (напр., систему масло — вода) амфи-фильного вещества [а в нек-рых случаях — спирта и (или) неорганич. соли] качественно изменяет свойства системы. Растворимость масла в воде резко возрас-тает при концентрации амфифильного вещества, превышающей ККМ. Молекулы гидрофобного вещества располагаются в углеводородных ядрах мицелл, размеры к-рых при этом увеличиваются — мицеллы набухают. Такое увеличение растворимости гидрофобных веществ в полярных растворителях (или полярных веществ в жирном растворителе) с образованием агрегатов наз. солюбилизацией. Радиус набухших мицелл в нек-рых случаях может достигать неск. сотен ; мицеллярные Р. при этом наз. микроэмуль-сиями типа «масло в воде» (o/w). Способность мицелл набухать в масле обычно ограничена, и при достижении нек-рой критич. концентрации масло отслаивается в виде отд. фазы. На рис. 18 изображён участок фазовой диаграммы солюбилизированного масла; равновесию фазы масла и солюбилизированного в воде масла отвечает кривая солюбилизации ВС.
Рис. 18. Схематическое изображение фазовой диаграммы масла, солюбилизированного в воде: 1 — трёхфазная область (вода + масло + амфифил); S — однородная прозрачная фаза (масло, солюбилизированное в воде); 3 — двухфазная система (солюбилизированное в воде масло + масло). АВ — кривая помутнения раствора; ВС— кривая солюбилизации; Tиф — температура инверсии фаз.
Радиус набухших мицелл определяется площадями, приходящимися на полярную «головку» на поверхностях раздела вода — амфифил () и масло — амфифил (). Для неионных амфифильных молекул, размеры полярной и гидрофобной частей к-рых примерно равны, предельное значение радиуса кривизны R поверхности масло — вода определяется соотношением
где L — полная длина амфифильной молекулы. Величина, стоящая в знаменателе этого выражения, зависит от температуры и состава Р., и при нек-рых значениях этих параметров (при температуре инверсии фаз) кривизна поверхности мицеллы может менять знак, т. е. происходит обращение микроэмульсии:(«масло в воде» : «вода в масле»). Вблизи температуры инверсии фаз мутность Р. резко возрастает и может наблюдаться, напр., опалесценция критическая .В узком интервале температур вблизи температуры инверсии фаз (заштрихованная область на рис. 18) в Р. обнаруживаются агрегаты разл. формы и состава: цилиндры, одно- и многослойные липосомы, ламеллы. Наблюдения в оптический и электронный микроскопы, а также данные малоуглового рассеяния рентг. излучения показали, что может возникать своеобразное подобие надмолекулярной организации микроэмульсий в интервале масштабов от неск. десятков нм до неск. мкм. Возможность возникновения развитой поверхности масло — вода вблизи температуры инверсии фаз связана с резким уменьшением величины поверхностного натяжения на этой границе в присутствии амфифильного вещества. Прямые измерения показывают, что добавление амфифильного вещества [а возможно, и спирта и (или) неорганич. соли] может привести практически к полному исчезновению поверхностного натяжения (т. н. ультранизкое поверхностное натяжение).
Коллоидные растворы
К коллоидным растворам, в широком смысле, кроме мпцеллярных Р. (наз. также ассоциирующими коллоидами) относят разнообразные суспензии (взвеси) частиц, таких, как белковые глобулы, вирусы (см. Клеточные структуры), металлические и полимерные золи и т. п. Частицы в коллоидном Р. взаимодействуют посредством слабых сил: ван-дер-ваальсово притяжение конкурирует с отталкиванием заряженных поверхностей частиц (в условиях экранирования зарядов ионами Р.). В случае гидрофильных частиц (т. н. гидрофильные коллоиды), к к-рым относятся глинистые минералы, а также практически все водорастворимые вещества биол. происхождения, существ. роль играют т. н. гидратац. силы, обязанные искажению структуры растворителя вблизи поверхностей частиц (на расстоянии 1-3 нм).
С течением времени частицы, совершающие в коллоидном Р. броуновское движение ,слипаются — коагулируют. Скорость этого процесса сильно зависит от величины потенц. барьера на кривой потенц. энергии взаимодействия частиц в Р. (рис. 19), отделяющего состояние слипшихся частиц от состояния системы до коагуляции. Высота потенц. барьера уменьшается с ростом концентрации ионов Р. и обращается в нуль при нек-рой их критич. концентрации, зависящей от температуры, электрич. свойств поверхностей коллоидных частиц и зарядов ионов (кривая 2). При концентрациях ионов, больших критической, имеет место т. н. быстрая коагуляция, при меньших — медленная коагуляция с флуктуац. преодолением потенц. барьера.
Рис. 19. Потенциальная энергия двух коллоидных частиц, находящихся на расстоянии R друг от друга: 1 — малая концентрация ионов; 2 — концентрация ионов, соответствующая порогу коагуляции; з — большая концентрация ионов.
При коагуляции могут возникать как упорядоченные, так и неупорядоченные агрегаты. К первым относятся, напр., тактоиды, образующиеся в Р., содержащих кап-сиды вируса табачной мозаики, и по существу представляющие собой нематические фазы лиотропных жидких кристаллов. Упорядоченные кубические кристаллы возникают в Р., содержащих полимерные частицы (напр., шарики латекса диаметром 100-1000 нм). К неупорядоченным агрегатам относятся т. н. гели, к-рые образуются в коллоидных Р. разл. состава и представляют собой упругие твёрдые тела, имеющие трёхмерный каркас из слипшихся коллоидных частиц — цилиндров, пластинок и т. п. Пустоты в каркасе заполнены растворителем. Полимерные гели образованы макромолекулами, скреплёнными между собой к—л. хим. агентами (напр., резина, набухшая в бензине, представляет собой Р. нитей каучука, «сшитых» в нек-рых точках серными мостиками). Содержание растворителя в геле может увеличиваться (набухание) или уменьшаться (осушение) при изменении температуры или (и) ионной силы Р. Как правило, набухание гелей ограничено, а избыток жидкости отслаивается в отд. фазу.
Модуль упругости геля зависит от концентрации «сшивок» между отд. частицами (рис. 20), причём конечный модуль сдвига появляется при нек-рой критич. концентрации «сшивок». Процесс образования геля из жидкого коллоида наз. фазовым переходом золь — гель.
Рис. 20. Зависимость модуля упругости К геля от концентрации «сшивок» между полимерными модулями [по оси абсцисс — параметр надкри-тичности (р/рс — 1), где p — концентрация «сшивок», рс — её критич. значение].
Коллоидные Р. глинистых минералов, подобных монтмориллониту, обладают свойством тиксотропии, а именно: при механич. размешивании Р. представляет собой жидкость, а в состоянии покоя — гель. Трёхмерный каркас монтмориллонитовых гелей образован крис-таллич. алюмосиликатными пластинками (диаметром в неск. сотен нм, толщиной ок. 1 нм), несущими заряды — отрицательные на поверхностях и положительные на торцах. В геле соседние пластинки могут быть ориентированы как параллельно друг другу (т. н. плотные контакты; в этом случае расстояние между ними определяется балансом электростатических, ван-дер-ва-альсовых и гидратационных сил; рис. 21), так и перпендикулярно друг другу. В зависимости от содержания воды и разл. солей (глинистые минералы обладают свойством избират. связывания ионов из раствора, напр. ионов К + , Cs + , Ca ++ , Sr ++ и др.) относит. доля контактов двух типов изменяется, а с ней изменяются и реологич. свойства гелей.
Рис. 21. Зависимость расстояния d между параллельными алюмосиликатными пластинками монтмориллонита от концентрации NaCl.
Особенность мн. коллоидных растворах — наличие значит. времён релаксации неравновесных состояний: даже в сравнительно разбавленных суспензиях релаксац. процессы могут длиться неделями и месяцами.
- http://cyberpedia.su/7x6e.html
- http://zdamsam.ru/a61197.html
- http://www.calc.ru/Teorii-Rastvorov.html
- http://bourabai.kz/physics/3325.html