При изучении характера наследования различных признаков у человека описаны все известные типы наследования и все типы доминирования. Многие признаки наследуются моногенно, т.е. определяются одним геном и наследуются в соответствии с законами Менделя. Моногенных признаков описано более тысячи. Среди них есть как аутосомные, так и сцепленные с полом. Некоторые из них приведены ниже.
| Признаки: | |
| доминантные | рецессивные |
| Темные волосы | Светлые волосы |
| Курчавые волосы | Прямые волосы |
| Карие глаза | Голубые глаза |
| Глаукома | Норма |
| Полидактилия | Норма |
| Заячья губа и волчья пасть | Норма |
Моногенные болезни встречаются у 1-2% населения земного шара. Это очень много. Частота спорадических моногенных болезней отражает частоту спонтанного мутационного процесса. Среди них большую долю составляют болезни с биохимическим дефектом. Типичным примером является фенилкетонурия.
Это тяжелое наследственное заболевание, обусловленное мутацией одного гена, нарушающей нормальный цикл превращения фенилаланина. У больных эта аминокислота накапливается в клетках. Болезнь сопровождается выраженной неврологической симптоматикой (повышенной возбудимостью), микроцефалией (маленькая голова) и в итоге приводит к идиотии. Диагноз ставится биохимически. В настоящее время в родильных домах проводится стопроцентное скринирование новорожденных на фенилкетонурию. Болезнь излечима, если вовремя перевести ребенка на специальную диету, исключающую фенилаланин.
Еще один пример моногенной болезни — синдром Морфана, или болезнь “паучьих пальцев”. Доминантная мутация одного гена имеет сильный плейотропный эффект. Помимо усиленного роста конечностей (пальцев), у больных наблюдается астения, порок сердца, вывих хрусталика глаза и другие аномалии. Болезнь протекает на фоне повышенного интеллекта, в связи с чем ее называют “болезнью великих людей”. Ею болели, в частности, американский президент А. Линкольн и выдающийся скрипач Н. Паганини.
Многие наследственные болезни связаны с изменением структуры хромосом или их нормального количества, т.е. с хромосомными или геномными мутациями. Так, тяжелое наследственное заболевание у новорожденных, известное как “синдром кошачьего крика”, вызвано утратой (делецией) длинного плеча 5-й хромосомы. Эта мутация приводит к патологическому развитию гортани, что вызывает характерный плач ребенка. Болезнь несовместима с жизнью.
Широко известная болезнь Дауна является результатом присутствия в кариотипе лишней хромосомы из 21-й пары (трисомия по 21-й хромосоме). Причиной служит нерасхождение половых хромосом при образовании половых клеток у матери. В большинстве случаев появления у новорожденных лишней хромосомы возраст матери достигает, по крайней мере, 35 лет. Мониторинг частоты этого заболевания в районах с сильным загрязнением окружающей среды обнаружил существенное увеличение количества больных этим синдромом. Предполагается также влияние вирусной инфекции на организм матери в период созревания яйцеклетки.
Отдельную категорию наследственных болезней составляют синдромы, связанные с изменением нормального количества половых хромосом. Как и болезнь Дауна, они возникают при нарушении процесса расхождения хромосом в гаметогенезе у матери.
У человека, в отличие от дрозофилы и других животных, Y-хромосома играет большую роль в определении и развитии пола. При отсутствии ее в наборе с любым количеством Х-хромосом особь фенотипически будет женской, а ее присутствие определяет развитие в сторону мужского пола. В частности, особи мужского пола с хромосомным набором ХХY + 44А больны синдромом Клайнфельтера. Они характеризуются умственной отсталостью, непропорциональным ростом конечностей, очень маленькими семенниками, отсутствием сперматозоидов, ненормальным развитием молочных желез и другими патологическими признаками. Увеличение числа Х-хромосом в сочетании с одной Y-хромосомой не изменяет определение мужского пола, а лишь усиливает синдром Клайнфельтера. Впервые кариотип ХХYY был описан в 1962 г. у 15-летнего мальчика со значительной умственной отсталостью, евнухоидными пропорциями тела, с уменьшенными в размере яичками и оволосением по женскому типу. Подобные же признаки характерны для больных с кариотипом ХХХYY.
 Синдром Клайнфельтера (1) и синдром Тернера-Шерешевского (2)
Синдром Клайнфельтера (1) и синдром Тернера-Шерешевского (2)
Отсутствие одной из двух Х-хромосом в кариотипе женщины (ХО) вызывает развитие синдрома Тернера-Шерешевского. Больные женщины обычно низкорослы, менее 140 см, коренасты, со слабо развитыми молочными железами, имеют характерные крыловидные складки на шее. Как правило, они бесплодны из-за недоразвития половой системы. Чаще всего беременность при этом синдроме приводит к самопроизвольному аборту. Только около 2% больных женщин сохраняют беременность до конца.
Трисомия (ХХХ) или полисомия по Х-хромосоме у женщин часто вызывает заболевание, сходное с синдромом Тернера-Шерешевского.
Наследственные болезни, связанные с изменением числа Х-хромосом, диагносцируются цитологическим методом по количеству в клетках телец Барра или полового хроматина. В 1949 г. М. Барр и Ч. Бертрам, изучая интерфазные ядра нейронов у кошки, обнаружили в них интенсивно красящееся тельце. Оно присутствовало только в ядрах клеток самок. Оказалось, что оно встречается у многих животных и всегда связано с полом. Эта структура получила название полового хроматина, или тельца Барра. В ходе тщательного цитологического и цитогенетического анализа было установлено, что половой хроматин представляет собой одну из двух женских половых хромосом, находящуюся в состоянии сильной спирализации и потому неактивную. У женщин с синдромом Тернера-Шерешевского (кариотип ХО) не обнаруживается полового хроматина, так же как и у нормальных мужчин ХY. Нормальные женщины ХХ и аномальные мужчины имеют по одному тельцу Барра, а женщины ХХХ и мужчины ХХХY — по два и т.д.
Лица с наследственными заболеваниями обычно рождаются с большими физическими отклонениями, что позволяет рано диагносцировать болезнь. Но иногда заболевание не дает о себе знать месяцами и даже десятилетиями. Например, тяжелая наследственная болезнь, вызванная поражением центральной нервной системы — хорея Гентингтона — может проявиться только после 40 лет, и тогда ее носитель успевает оставить потомство. Для больных характерны непроизвольные подергивающиеся движения головы и конечностей.
Бывает так, что человек производит впечатление абсолютно здорового индивидуума, но у него есть наследственная предрасположенность к определенному заболеванию, которое проявляется под воздействием внешних или внутренних факторов. Например, некоторым людям свойственна тяжелая реакция на определенные лекарственные препараты, которая обусловлена генетическим дефектом — отсутствием в организме специфического фермента. Иногда наблюдается смертельная реакция на наркоз с виду совершенно здоровых людей, но на самом деле носящих в себе в скрытом виде особую наследственную болезнь мышц. У таких пациентов во время или после операции, проходящей под наркозом, внезапно подскакивает температура (до 42°).
Читайте также другие статьи темы 12 «Генетика человека»:
Перейти к чтению других тем книги «Генетика и селекция. Теория. Задания. Ответы»:
Молекулярные бомбы с программным управлением
Виола Брик, «Вокруг Света»
Когда говорят о «плохих» или «хороших» генах, подразумевают, как правило, очевидные качества человека. Но многие гены никак себя не проявляют. Об их существовании мы узнаем в самый неподходящий момент. Они выгодны эволюции, но остаются головоломкой для медицины.
Гонка за геномом
Организм человека формируется и развивается далеко не случайным образом. Все проходящие в нем процессы подчиняются определенному плану, записанному на довольно сложном языке – в виде линейной последовательности пар оснований молекулы ДНК. Ещё пятнадцать лет назад этот язык был загадкой для науки. В 1990 году в США стартовала межгосударственная программа «Геном человека» (Human Genome Project). В проекте участвовали государственные лаборатории восемнадцати стран мира, и им удалось к 2003 году практически полностью расшифровать геном человека. Интересно, что подобные исследования проводила и частная компания «Celera genomics», в планы руководства которой входило оформление патента на каждый открытый ген и, как следствие, получение прибыли за предоставление информации. Конкурирующие организации по взаимной договорённости одновременно заявили о завершении работ. 26 июня 2000 года было объявлено, что геном человека расшифрован на 97%. Сегодня исследования перешли в иную область: от структурной геномики к геномике функциональной, которая поможет установить, как управляются и работают гены.
Большая часть генома человека сосредоточена в ДНК клеточных ядер. Этот генетический материал организован в парный набор хромосом. Половину хромосом человек получает от матери, вторую половину – от отца. При этом каждый внешний признак, будь то цвет волос или оттенок кожи, определяется двумя генами (по одному от каждого родителя). В зависимости от того, какой ген доминирует, ребенок становится в чём-то похожим либо на отца, либо на мать. Получается, что для передачи важных признаков гены должны быть надежно защищены. Действительно, здоровый геном постоянно «проверяется» специальными ферментами, которые идентифицируют и устраняют неполадки. Но такой механизм не всегда эффективен, поэтому время от времени в геноме происходят мутации – модификации того или иного участка в структуре гена, например, его удлинение, укорочение, изменение смысла. Мутации могут происходить либо спонтанно, либо при агрессивных воздействиях, например таких, как ультрафиолетовое излучение. С эволюционной точки зрения спонтанные мутации очень выгодны, ведь они корректируют показатели живого организма, обеспечивая его приспособление к изменяющимся условиям окружающей среды и в конечном итоге появление новых видов животных и растений. Но возможен и другой вариант, когда мутация оказывается опасной для организма, и, передаваясь потомкам, таит в себе болезнь.
Генетическими называют те заболевания, причина которых не в инфекции и не в случайных сбоях в работе организма, а в самом «плане», заложенном в молекуле ДНК. Человек получает болезнь от родителей, если в их генах произошла мутация. Статистика неутешительна: примерно 70% населения планеты несет в себе геном с теми или иными отклонениями от нормы. Однако частота проявления подобных нарушений не столь велика. Дело в том, что для генетических нарушений важно не только присутствие модифицированного участка ДНК, но и его передача последующим поколениям.
Генетические заболевания различаются по механизму возникновения и наследования, среди них выделяют менделевские, хромосомные, мультифакторные и другие. Менделевские болезни наследуются в соответствии с законами Менделя (Gregor Johann Mendel, 1822–1884), согласно которым в потомстве проявляются доминирующие признаки родителей. Большинство генетических мутаций не доминируют, остаются «молчащими» на протяжении всей жизни человека. Только если и мать, и отец являются носителями измененного гена, ребенок унаследует ген-мутант. Именно поэтому браки между членами одной семьи, а также внутри ограниченной группы людей зачастую приводят к рождению потомства с генетическими отклонениями. К группе менделевских относят и заболевания, связанные с половыми хромосомами Х и У. Например, синдром fra-X (синдром хрупкой Х хромосомы), или синдром Мартина-Белл, встречается преимущественно у мальчиков. В основе болезни лежат изменения в гене FMR-1, связаные с увеличением числа копий тринуклеотидного повтора CGG (цитидин-гуанин-гуанин). В норме такая последовательность составляющих ДНК нуклеотидов не превышает 50–54, в то время как в ДНК больного человека встречается до полутора тысяч повторов.
К началу третьего тысячелетия ученым было известно около одиннадцати тысяч менделевских наследственных заболеваний, и их число постоянно растёт. На сегодняшний день разгаданы и молекулярные механизмы многих дефектов. Так, при анализе семисот шестидесяти семи дефектных генов человека было обнаружено, что из них шестьсот пятьдесят восемь повинны в возникновении только одного нарушения, семьдесят один бракованный ген отвечает за два нарушения, тридцать – за три. Существуют бракованные гены, которые приводят к развитию и пяти, шести или семи нарушений. Одна коварная мутация в одном-единственном гене может привести к нарушению целого ряда функций! Та же хрупкая Х хромосома приводит не только к умственной отсталости, но и часто характеризуется близорукостью, «заячьей губой», апноэ, сколиозом и сердечно-сосудистыми нарушениями.
Нарушения могут наблюдаться не только в микроструктуре генов, как это происходит с менделевскими болезнями, но и на макроуровне. Такие отклонения называются хромомсомными. К примеру, синдром Дауна (John Langdon Down, 1828–1896) – одно из самых распространенных нарушений умственного развития – связан с тем, что новорожденный получает три хромосомы номер 21 вместо двух. Впрочем, в 5–8% случаев аномалия связана не с лишней, третьей, хромосомой, а с её фрагментом. В 21-й хромосоме расположен ген, ответственный за выработку миоинозитола, избыток которого и вызывает умственную отсталость. В настоящее время учёные пытаются найти способ нормализовать концентрацию этого вещества в организме больных. Но всё же возможность медикаментозного лечения синдрома Дауна представится человечеству не скоро.
До недавнего времени были изучены преимущественно моногенные, то есть возникающие при нарушении работы одного гена, заболевания. Но большинство наследственных болезней связаны с одновременным нарушением работы нескольких генов и определённым воздействием внешней среды. Такие болезни называют мультифакторными. Например, обнаружено несколько генов, мутации которых связывают с проявлением болезни Альцгеймера. Это ген предшественника амилоидного белка (АРР) на хромосоме 21, ген аполипопротеина Е (АроЕ) на хромосоме 19, ген пресенилина-1 на хромосоме 14 и ген пресенилина-2 на хромосоме 1. Болезнь Альцгеймера (Alois Alzheimer, 1864–1915), сахарный диабет, эпилепсия, сердечная недостаточность, астма, шизофрения вызваны не только «испорченным» геномом, но и неблагоприятными факторами внешней среды.
Частота проявлений мультифакторных заболеваний среди родственников заметно выше, чем среди неродственных особей, однако первопричина до сих пор остается загадкой.
Многие генетические болезни крайне редки. Сегодня известно несколько сотен нарушений, которыми страдают лишь единицы из миллионов, например, нарушение толщины кожи или костей, некоторые расстройства психики. Есть среди генетических нарушений действительно экзотические, такие как синдром преждевременного старения у детей, или синдром Хатчинсона-Гилфорда (Jonathan Hutchinson, 1828–1913; Hastings Gilford, 1861–1941). Причина его – в генетической мутации, из-за которой в клетках накапливается аномальный протеин, что приводит к деформации ядра клетки. Нестабильность ядерных мембран вызывает изменения в тканях и ускоряет смерть клеток. Симптомами болезни являются карликовость, облысение, появление морщин, уплотнение стенок артерий и остеопороз. Продолжительность жизни детей, страдающих этой редкой болезнью, не превышает 15–17 лет.
А вот синдром Жиля де ля Туретта (Gilles de la Tourette, 1857–1904) не угрожает жизни, зато связан с дискомфортом как больного, так и окружающих. При этом синдроме время от времени происходят непроизвольные движения мышц и мозговая активность, что проявляется в подергивании рук, ног и сопровождается выкрикиванием ругательств.
Пожалуй, самым редким заболеванием можно назвать недавно открытый синдром «холодного пота», от которого страдают всего несколько человек в мире. При этом заболевании потоотделение происходит при низких температурах, а не на жаре, как у большинства людей. Причины этого синдрома пока неизвестны, но могут быть связаны с генетическими нарушениями в развитии суставной ткани.
Следует отметить, что редкие генетические болезни очень сложны в изучении. Несколько десятков человек во всем мире – недостаточная статистика для проведения научных исследований. Кроме того, финансирующие организации неохотно выделяют средства на изучение редких мутаций, поскольку более распространённые заболевания приносят больше вреда, и изучение их более привлекательно для фармацевтической индустрии.
На сегодняшний день установлено несколько тысяч генов, которые вызывают или обуславливают предрасположенность к развитию заболеваний. Во всем мире уже разработаны тесты, позволяющие выявить более пятисот различных болезней. Так, ещё в утробе матери можно определить наличие или оценить риск появления многих страшных болезней, включая и синдром Дауна. Пары, планирующие ребенка, тоже могут пройти генетическое тестирование на предмет хранения «молчащих» дефектных генов, которые могут проявить себя в потомстве. Правда, генетическая диагностика остается дорогостоящей процедурой, поэтому скрининг обычно сводится лишь к нескольким десяткам самых распространенных заболеваний. Врачи либо выявляют заболевание ещё в утробе, либо дают знать, что риск развития болезни после рождения ребёнка очень велик. В таких случаях рекомендуют аборт.
Кстати, многие медики сходятся во мнении, что выкидыши могут быть связаны с генетически неполноценным плодом. Таким образом организм избавляется от нежизнеспособного эмбриона, либо сам эмбрион не развивается полноценно, что приводит к абортации. Если у женщины было уже несколько выкидышей, то при очередной беременности врачи настаивают на пренатальной диагностике: в таких случаях велика вероятность того, что родители передают эмбрионам тяжелую генетическую болезнь, что и приводит к частым выкидышам.
Но даже для носителей дефектных генов остается надежда. Генная терапия – одно из самых перспективных направлений современной медицины. История генной терапии началась в 1990 году, когда американский генетик Уильям Андерсон (William French Anderson) впервые применил этот метод на маленькой пациентке – четырехлетняя девочке с иммунодефицитом, вызванным недостаточной функцией фермента аденозиндеаминазы (АДА). Врач ввел в организм девочки вирус с геном, кодирующим этот фермент. Клетки пациентки стали считывать вирусный геном и восполнили недостаток АДА. Хотя процедура закончилась успешно, генная терапия пока не стала массовой. Дело в том, что геном каждого пациента уникален, поэтому к каждому требуется персональный подход. К тому же геном человека постоянно мутирует. В настоящее время в мире проводится множество клинических испытаний по генной терапии тех или иных заболеваний, что позволяет человечеству не терять надежду в борьбе с тысячами недугов.
Какие болезни передаются по наследству — список, классификация, генетические тесты и профилактика
Человек за время своей жизни переносит множество легких или тяжелых болезней, но в некоторых случаях он рождается уже с ними. Наследственные заболевания или генетические нарушения проявляются у ребенка из-за мутации одной из хромосом ДНК, что приводит к развитию недуга. Некоторые из них несут лишь внешние изменения, но существует ряд патологий, которые угрожают жизни малыша.
Что такое наследственные заболевания
Это генетические болезни или хромосомные аномалии, развитие которых связано с нарушением в наследственном аппарате клеток, передающиеся через репродуктивные клетки (гаметы). Возникновение таких наследственных патологий связано с процессом передачи, реализации, хранения генетической информации. Все больше мужчин имеют проблему с отклонениями такого рода, поэтому шанс зачать здорового ребенка становится все меньше. Медицина ведет постоянные исследования для выработки процедуры предотвращения рождения детей с отклонениями.



Генетические заболевания наследственного типа формируется при мутации генной информации. Выявлены они могут быть сразу же после рождения ребенка или, спустя длительное время при долгом развитии патологии. Выделяют три главные причины развития наследственных недугов:
- хромосомные аномалии;
- нарушения хромосом;
- генные мутации.
Последняя причина входит в группу наследственно предрасположенного типа, потому что на их развитие и активизацию влияют еще и факторы внешней среды. Ярким примером таких заболеваний считается гипертоническая болезнь или сахарный диабет. Кроме мутаций на их прогрессирование влияет длительное перенапряжение нервной системы, неправильное питание, психические травмы и ожирение.

Каждая наследственная болезнь имеет свои специфические признаки. На данный момент известно свыше 1600 разных патологий, которые становятся причиной генетических и хромосомных аномалий. Проявления отличаются по степени тяжести и яркости. Для предотвращения появления симптомов необходимо вовремя выявить вероятность их появления. Для этого используют следующие методы:
- Близнецовый. Наследственные патологии диагностируются при изучении различия, сходства близнецов для определения влияние генетических особенностей, внешней среды на развитие заболеваний.
- Генеалогический. Вероятность развития патологических или нормальных признаков изучается при помощи родословной человека.
- Цитогенетический. Исследуются хромосомы здоровых и больных людей.
- Биохимический. Проводится наблюдение за обменом веществ у человека, выделяются особенности этого процесса.
Помимо этих методов большинство девушек во время вынашивания ребенка проходят ультразвуковое исследование. Оно помогает определить по признакам плода вероятность появления врожденных пороков развития (с 1 триместра), предположить присутствие у будущего ребенка определенного ряда хромосомных болезней или наследственных недугов нервной системы.


Подавляющее большинство заболеваний наследственного характера проявляются еще в детстве. Каждая из патологий имеет собственные признаки, которые уникальны для каждой болезни. Аномалий большое количество, поэтому подробнее они будут описаны ниже. Благодаря современным методам диагностики выявить отклонения в развитии ребенка, определить вероятность наследственных болезней можно еще во время вынашивания ребенка.
Классификация наследственных болезней человека
Объединение в группы заболеваний генетического характера проводится по причине их возникновения. Основными видами заболеваний наследственного характера являются:
- Генетические – возникают из повреждений ДНК на уровне гена.
- Предрасположенность по наследственному типу, аутосомно-рецессивные заболевания.
- Хромосомные аномалии. Заболевания возникают вследствие появления лишней или утери одной из хромосом или их аберрациями, делеции.
Список наследственных заболеваний человека
Науке известно более 1500 болезней, которые относятся к вышеописанным категориям. Некоторые из них встречаются крайне редко, но определенные виды на слуху у многих. К самым известным относятся следующие патологии:
- болезнь Олбрайта;
- ихтиоз;
- талассемия;
- синдром Марфана;
- отосклероз;
- пароксизмальная миоплегия;
- гемофилия;
- болезнь Фабри;
- мышечная дистрофия;
- синдром Клайнфельтера;
- синдром Дауна;
- синдром Шерешевского-Тернера;
- синдром кошачьего крика;
- шизофрения;
- врожденный вывих бедра;
- пороки сердца;
- расщепление неба и губы;
- синдактилия (срастание пальцев).
Какие наиболее опасны
Из вышеперечисленных выше патологий существуют те болезни, которые считается опасными для жизни человека. Как правило, входит в этот список те аномалии, которые имеют в хромосомном наборе полисомию или трисомию, когда вместо двух наблюдается от 3 до 5 или больше. В некоторых случаях обнаруживается 1 хромосома вместо 2. Все такие аномалии становятся следствие отклонений при делении клеток. При такой патологии ребенок живёт до 2 лет, если отклонения не очень серьезные, то доживает до 14 лет. Самыми опасными недугами считаются:
- болезнь Кэнэвэн;
- синдром Эдвардса;
- гемофилия;
- синдром Патау;
- спинальная мышечная амиотрофия.
Синдром Дауна
Болезнь передается по наследству, когда оба или один из родителей имеет дефектные хромосомы. Синдром Дауна развивается из-за трисомии21 хромосомы (вместо 2 находится 3). дети с этим недугом страдают косоглазием, имеют аномальную форму ушей, складку на шее, наблюдается умственная отсталость и проблемы с сердцем. Опасности для жизни эта аномалия хромосом не представляет. По статистике из 800 рождается 1 с данным синдромом. Женщины, которые хотят родить после 35 вероятность рождения ребенка с Дауном вырастает (1 к 375), после 45 лет вероятность 1 к 30.

Акрокраниодисфалангия
Недуг имеет аутосомно-доминантый тип наследования аномалии, причиной становится нарушение в 10 хромосоме. Ученые называют болезнь акрокраниодисфалангия или синдром Аперта. Характеризуется следующими симптомами:
- нарушения соотношения длины и ширины черепа (брахикефалия);
- внутри черепа формируется повышенное кровяное давление (гипертензия) по причине срастания коронарных швов;
- синдактилия;
- умственная отсталость на фоне сдавливания мозга черепом;
- выпуклый лоб.
Каковы возможности лечения наследственных заболеваний
Врачи постоянно работают над проблемой аномалий генов и хромосом, но все лечение на данном этапе сводится к подавлению симптомов, полного выздоровления добиться не удается. Подбирается терапия в зависимости от патологии, чтобы снизить выраженность признаков. Часто используют применяют такие варианты лечения:
- Увеличение количества поступающих коферментов, к примеру, витаминов.
- Диетотерапия. Важный пункт, который помогает избавиться от целого ряда неприятных последствий наследственных аномалий. При нарушении диеты сразу наблюдается резкое ухудшение состояние больного. К примеру, при фенилкетонурии полностью из рациона исключается продукты, которые содержат фенилаланин. Отказ от этой меры может привести к тяжелой идиотии, поэтому врачи акцентируют внимание на необходимости диетотерапии.
- Потребление тех веществ, которые отсутствуют в организме по причине развития патологии. К примеру, при оротацидурии назначает цитидиловую кислоту.
- При нарушении обмена веществ необходимо обеспечить своевременное очищение организма от токсинов. Болезнь Вильсона-Коновалова (накопление меди) купируется приемом д-пеницилламина, а гемоглобинопатия (накопление железа) – десфералом.
- Ингибиторы помогают блокировать чрезмерную активность ферментов.
- Возможна трансплантация органов, участков ткани, клеток, которые содержат нормальную генетическую информацию.

Профилактика
Специальные анализы помогают определить вероятность появления заболевания наследственного типа еще во время беременности. Для этого применяют молекулярно-генетическое исследование, которое несет некоторый риск, поэтому перед его выполнением следует обязательно посоветоваться с врачом. Профилактика наследственных заболеваний проводится только при условии, что женщина находится в группе риска и есть возможность наследования нарушений ДНК (к примеру, всем девушкам после 35 лет).
Наследственные заболевания
На сегодняшний день известно больше четырёх с половиной тысяч наследственных заболеваний, и каждый случай имеет твердую доказательную базу о том, что болезнь передается именно по наследству, и никак иначе. Но, несмотря на высокий уровень развития диагностики, далеко не все генетические патологии изучены до степени биохимических реакций. Тем не менее, основные механизмы развития наследственных заболеваний известны современной науке.
Существует три базовых вида мутаций:
- Генные;
- Хромосомные;
- Геномные (преимущественно сцепленные с полом).
Основополагающие генетические законы Менделя определяют доминантные и рецессивные гены. После оплодотворения клетки плода содержат половину генов матери и половину отца, составляя пары – аллели. Генетических комбинаций не так много: всего две. Определяющие признаки проявляются в фенотипе. Если один из мутировавших генов аллели доминантен – болезнь проявляется. То же самое происходит при доминантной паре. Если такой ген рецессивный, в фенотипе это никак не отражается. Проявление наследственных заболеваний, передающихся по рецессивному признаку, возможно только при условии, что оба гена несут патологическую информацию.
Хромосомные мутации проявляются нарушением их деления в процессе мейоза. В результате дупликации появляются дополнительные хромосомы: как половые, так и соматические.
Сцепленные с полом наследственные аномалии передаются посредством половой Х-хромосомы. Поскольку у мужчин она представлена в единственном числе, у всех лиц мужского пола в роду имеются проявления болезни. Тогда как женщины, имеющие две половых Х-хромосомы, являются носителями поврежденной Х-хромосомы. Для проявлений у женщин наследственного заболевания, сцепленного с полом, необходимо, чтобы пациентка унаследовала обе дефектные половые хромосомы. Это случается достаточно редко.
Биология наследственных заболеваний
Проявления наследственной патологии зависят от многих факторов. Признаки, заложенные в генотипе, имеют внешние проявления (затрагивают фенотип) при определенных условиях. В связи с этим биология наследственных заболеваний разделяет все генетически обусловленные болезни на следующие группы:
- Проявления, не зависящие от внешней среды, воспитания, социальных условий, благосостояния: фенилкетонурия, болезнь Дауна, гемофилия, мутации половых хромосом;
- Наследственная предрасположенность, которая проявляется только при определенных условиях. Большое значение имеют факторы внешней среды: характер питания, профессиональные вредности и пр. К таким заболеваниям относятся: подагра, атеросклероз, язвенная болезнь, артериальная гипертензия, сахарный диабет, алкоголизм, опухолевый рост клеток.
Иногда, признаки даже ненаследуемых заболеваний обнаруживаются у детей больных людей. Этому способствует одинаковая восприимчивость у родственников к определенным факторам. Например, развитие ревматизма, возбудитель которого не имеет никакого отношения к генам и хромосомам. Тем не менее дети, внуки и правнуки также подвержены системному поражению соединительной ткани β-гемолитическим стрептококком. Многих людей хронический тонзиллит сопровождает всю жизнь, но наследственных заболеваний не вызывает, тогда как у имеющих родственников с ревматическими поражениями клапанов сердца, развивается аналогичная патология.
Причины наследственных заболеваний
Причины наследственных заболеваний, связанных с генными мутациями, всегда одинаковы: дефект гена – дефект фермента – отсутствие синтеза белка. В результате в организме скапливаются вещества, которые должны были превратиться в необходимые элементы, но сами по себе как промежуточные продукты биохимических реакций являются токсичными.
Например, классическое наследственное заболевание – фенилкетонурия обусловлено дефектом гена, который регулирует синтез фермента, превращающего фенилаланин в тирозин. Поэтому при фенилкетонурии страдает головной мозг.
При недостаточности лактазы возникает расстройство кишечника. Непереносимость сырого коровьего молока – явление достаточно распространённое, и оно также относится к наследственным заболеваниям, хотя, при определенных условиях, у некоторых людей может произойти компенсация, и выработка лактазы налаживается в связи с активной «тренировкой» клеток кишечника.
Хромосомные аномалии проявляются независимо от условий. Многие дети оказываются просто нежизнеспособными. Но болезнь Дауна относится к тем наследственным заболеваниям, при которых внешние условия среды могут оказаться настолько благоприятными, что больные становятся полноправными членами общества.
Дефекты деления половых хромосом не сопровождаются смертельными осложнениями, так как не задевают соматических признаков. Все жизненно важные органы при таких наследственных заболеваниях не страдают. Повреждения обнаруживаются на уровне половых органов, причем часто только внутренних. Иногда обходится и без них. Например, при синдроме трипло-экс, когда у женщины имеется добавочная Х-хромосома, способность к зачатию сохраняется. И дети рождаются с нормальным набором половых хромосом. Аналогичная ситуация с дополнительной Y-хромосомой у мужчин.
Механизм развития наследственных заболеваний кроется в сочетании генов: доминантных и рецессивных. Различное их сочетание неодинаково проявляется в фенотипе. Для развития заболевания достаточно одного мутировавшего доминантного гена, либо патологической рецессивной пары в одном аллеле.
Профилактика наследственных заболеваний
Предотвращением проявлений генетической патологии занимаются специалисты генетических центров. В женских консультациях крупных городов работают специальные кабинеты генетиков, которые проводят консультирование будущих семейных пар. Проводится профилактика наследственных заболеваний путем составления генеалогических карт и расшифровки специальных анализов.
Наследственные заболевания
К биологическим видам неприменимо понятие неизменности. С каждым новым поколением особи наследуют новые гены — мутируют. Изредка мутация может улучшить шансы индивидуума на выживание, и тогда этот новый ген широко распространяется во всей популяции. Подавляющее большинство мутаций, однако, приводит к сокращению потомства. Это и есть классическое равновесие мутации и смерти, называемое естественным отбором и принятое биологами как бесспорное для всех видов.
В этой книге рассмотрены некоторые философские вопросы, касающиеся ценностей и целей человеческой цивилизации. Нас интересует путь, по которому будет развиваться человечество, сознательно выбрав одно из двух: применять или отвергнуть искусственный отбор.
Книга эта была задумана отнюдь не с целью принять участие в дискуссии о сложностях генетической патологии человека. Если подыскивать аналогии, наше изложение можно сравнить скорее с дорожной картой, нежели с руководством по ремонту автомобиля. Но поговорить о некоторых гайках и болтах все же придется»
Успехи медицины таковы, что естественный отбор свелся почти к нулю; уже 98% американцев доживают по меньшей мере до 25 лет[20]. Медицина заботится преимущественно о ныне живущих людях. В частности, упор делается на инфекционные заболевания, передаваемые «горизонтально», а не на генетические заболевания, распространяющиеся «по вертикали». В конце концов, врачу и фармацевту очень сложно взимать плату с еще не рожденных людей. Медицина в нашем обществе — бизнес, ориентированный на платных пациентов; а ведь те, кто может и хочет платить, — это люди, которые больны сейчас.
Британская энциклопедия кратко перечисляет некоторые из характерных фактов, относящихся к 3500 известным в настоящее время аутосомным доминантным и рецессивным заболеваниям, а также заболеваниям, сцепленным с полом (список этот, впрочем, постоянно растет):
Эпидемиологические исследования дают основания предполагать, что примерно 1 процент всех новорожденных страдает одним генным дефектом и 0,5 процента имеют хромосомные аномалии, способные привести к серьезным физическим дефектам и умственной отсталости. Исследования показывают, что по меньшей мере половина из 3—4 процентов младенцев с врожденными дефектами несет в себе значительные генетические недостатки. Как минимум пять процентов всех регистрируемых зачатий имеют серьезные хромосомные аномалии, а в сорока—пятидесяти процентах самопроизвольных абортов речь идет об эмбрионах с хромосомными аномалиями. Около сорока процентов детской смертности обусловлено генетической патологией; тридцать процентов детских и десять процентов взрослых пациентов нуждаются в больничном уходе из-за генетических расстройств. Специалисты подчеркивают, что генетические дефекты, хотя бы и небольшие, имеются у десяти процентов всех взрослых… Пятая часть мертворождений и смертей в раннем детском возрасте вызвана серьезными врожденными аномалиями, и около семи процентов всех новорожденных страдают от умственных или физических дефектов.
Это далеко не все. Показатели спонтанных мутаций свидетельствуют, что такого рода генетических «опечаток» ныне приходится до ДВУХСОТ на каждого человека[22]. По большей части они безобидны, однако неизвестно, какой процент окажется нежелательным при экспрессии, учитывая их кумулятивный эффект. Помимо генетических аномалий, необходимых и достаточных для появления соответствующих болезней, существует намного больше многофакторных заболеваний, в которых соучаствуют определенные гены, создавая ту или иную степень предрасположенности к конкретным заболеваниям, например к большинству разновидностей рака, сахарному диабету, гипертонии.
Ранние евгеники наивно полагали, что достаточно будет просто не позволять людям с наследственными заболеваниями иметь детей, чтобы с каждым поколением появлялось все больше здоровых людей. Но патологические гены чаще всего рецессивны и крайне редки. Таким образом, число носителей нежелательных генов намного превышает количество активно больных, и отказ больных людей производить потомство мог бы дать лишь крайне медленный спад заболеваемости в последующих поколениях. Например, если та или иная наследственная патология проявила себя у одного процента населения, потребуется ДЕВЯНОСТО поколений, чтобы снизить этот показатель до одной сотой процента, и ДЕВЯТЬСОТ поколений — при условии произвольного спаривания, — чтобы понизить этот показатель до одного случая на миллион[23]. Но и тогда естественные спонтанные мутации будут продолжаться, и воевать с ними придется до бесконечности.
Генная инженерия быстро развивается. В случае, если один или оба будущих родителя являются носителями генетических заболеваний, врач может осуществить искусственное осеменение in vitro и затем провести эмбриональный скрининг — доимплантационную генетическую диагностику, чтобы выбрать здоровый эмбрион для имплантации в матку. Подобные евгенические мероприятия уже теперь предпринимаются на добровольной основе. В недалеком будущем станет возможным вносить изменения в эмбриональные, а не только соматические (не участвующие в размножении) клетки. Терапия зародышевого пути на самом деле не относится ни к позитивной, ни к негативной евгенике. Но это евгеника. Когда впервые появились возможности такого рода, евгеническая наука встретила полное и безоговорочное осуждение; ныне разговор идет по большей части об установлении моратория на эту новую терапию. Фриц Манн, специалист по биологической этике, работающий в Свободном университете Брюсселя, пишет:
Помимо религиозных причин, никаких этических оправданий для отказа от влияния на зародышевый путь не существует, И если откроют способ излечения наследственной болезни, причем излечения не только самого носителя, но и всех его потомков, то с какой стати его запрещать?
Такое открытие ознаменует прорыв в генетике, но загадка генов и их взаимодействий еще ждет окончательного решения. Тем не менее генетики уже меняют наследственные механизмы у животных и растений, и терапия зародышевого пути в человеческих популяциях — лишь вопрос времени. Сейчас генетическая консультация и соответствующее лечение в некоторых случаях помогают ныне живущим принести пользу потомству. Будущий родитель, зная, что он (или она) является носителем рецессивного гена, способного вызвать заболевание в последующих поколениях, может абортировать зародыш. Таким образом, дети избавляются от этой болезни, но число носителей рецессивного гена возрастает с каждым звеном в цепочке поколений.
Вопрос заключается в следующем: имеют ли родители моральное право производить на свет детей с нездоровой наследственностью? Философа теолог Эммануил Левинас формулирует это следующим образом: «Мой сын — не просто мое создание, как стихотворение или вещь. Он — не моя собственность»[25]. Можно ли отбрасывать, отрицать родительскую ответственность? Профессор Института детского здоровья при Лондонском университете Маркус Пембрей, говоря о генетической консультации, утверждает:
Целью не должно быть сокращение числа новорожденных с генетическими заболеваниями, ибо это означало бы обойти право матери делать или не делать аборт… Точка зрения, согласно которой сокращение числа младенцев, рожденных с генетическими расстройствами, не является подходящей задачей для генетических служб, находит сейчас широкий отклик.
Это и есть так называемая «модель личного обслуживания»[27], которая подчиняет благополучие детей воле их родителей. Такая точка зрения прямо-таки просится на судебное обжалование на основании «неправомерной жизни» (по аналогии с процессами, которые впервые появились в Соединенных Штатах в 1964 г., утвердив иски по «неправомерной смерти» в качестве законного прецедента). Прежде нам не хватало знаний для борьбы с наследственными болезнями; в будущем ссылки на неосведомленность будут все меньше приниматься во внимание. Такую политику невозможно будет сравнивать, например, со скандалом в связи с применением талидомида в 1957—1961 гг., поскольку речь будет идти о действиях, совершенных осознанно и преднамеренно.
Вмешательство в зародышевый путь столкнется с сопротивлением тех, кто считает (в частности, по религиозным соображениям) подобную терапию противоестественной: по их мнению, мы не имеем права «играть роль Бога». Отдельные религиозные группы отвергают вообще всякое лечение. Время от времени в газетах появляются сообщения о семьях, где ребенок умер из-за отказа родителей от медицинского ухода. Возникнут и нерелигиозные возражения со стороны людей, которые опасаются медицинских ошибок. Надо признать, что ошибки в самом деле возможны. Но по мере накопления знаний в области человеческой генетики аргументы оппонентов утратят силу.
Ведущую роль во внедрении генетических консультаций играет государство Израиль. Вот что пишет Гидеон Бах, декан факультета генетики Еврейского университета в Иерусалиме:
Сейчас мы знаем, что если не все, то большинство болезней человека имеют генетические предпосылки, и овладеваем методами изучения, лечения и, в конечном счете, предотвращения этих болезней… Израиль, с его многочисленными этническими группами, внутри которых дети нередко рождаются от кровных родственников, оказался богатой лабораторией для исследователей генетики. Наследственные аномалии гораздо легче проследить в группах с гомогенетическими родословными.
Евреи Восточной Европы, которые столетиями, если не считать последних сорока лет, вступали в преимущественно родственные браки, относительно часто оказываются носителями десятка рецессивных генетических заболеваний. Наиболее известно аутосоматическое расстройство, описанное в 1881 году британским офтальмологом Уорреном Тэем и названное болезнью Тэя-Сакса. Оно вызвано наследственной утратой необходимого фермента, который обычно разрушает жировые отложения в мозгу. Если оба родителя являются носителями этого гена, ребенок в двадцати пяти случаях из ста страдает этой болезнью, в пятидесяти из ста становится ее носителем (кондуктором). Один из двадцати семи евреев в Соединенных Штатах — носитель этого гена. Больной младенец сначала кажется нормальным, но через несколько месяцев становится сверхчувствительным к звуку. В конце концов ребенок глохнет, слепнет, становится умственно отсталым и невосприимчивым к внешним раздражителям и умирает примерно в пятилетнем возрасте.
В 1985 году раввин Иосиф Экштейн, опираясь на Библию и Талмуд, основал международную генетическую программу тестирования под названием «Дор иешорим» («поколения праведных»), цель которой — уберечь будущих детей от врожденных заболеваний. По этой программе студенты — ортодоксальные евреи — подвергаются тестированию, чтобы определить, не являются ли они носителями данного гена. Если один из будущих супругов оказывается носителем, их не отговаривают от брака, если же тест положителен у обоих, им советуют выбрать другого брачного партнера.
В Израиле реализована одна из самых интенсивных программ генетического скрининга, тестам подвергается более десяти тысяч человек в год[29]. Писательница Наоми Стоун выразила общее отношение евреев к профилактике болезни Тэя-Сакса:
Не исключено, что отдельную болезнь можно искоренить в той части населения, где сконцентрированы носители патологического гена, и если бы это удалось, кто мог бы всерьез усомниться в целесообразности подобных мер. Я — еврейка-ашкенази, и знаю, что мой долг всегда быть начеку из-за повышенного риска.
Не приходится удивляться тому, что в Соединенных Штатах, напротив, решительно возражают против использования евгеники. Так обстоит дело, например, у представителей общества инвалидов. Биоэтик Эйдриен Эш пишет:
Мое моральное неприятие предродового тестирования и выборочных абортов исходит из убеждения, что жизнь и с ограниченными возможностями несет в себе собственное оправдание. Я убежден, что любое справедливое общество обязано ценить и охранять жизнь всех людей, независимо от того, какие способности им выпали в лотерее природы.
Во многом сходной позиции придерживается канадский ученый-этик Том Коч, доказывающий, что все болезни — это часть многообразия человеческой расы[32].
Грегор Уолбринг, еще один канадец, активист движения инвалидов против евгеники, родившийся без ног, и отец дочери, страдающей синдромом Дауна, идет еще дальше:
Могу сказать без колебаний, что мой недостаток обогатил мою жизнь. Как может тот, кто не испытал его, это понять?
Мистер Уолбринг основал веб-сайт с материалами сторонников и противников евгеники[34]. Сам он при этом — ее противник.
А вот что можно прочесть в другом документе, распространенном через Интернет:
Главное в обсуждении евгеники — это то, что кто-то, основываясь на гласных или негласных критериях, берет на себя право решать, какие свойства и способности человека имеют право на существование, а какие — нет. Ключевой вопрос — как общество (социальная евгеника) или личность (личная евгеника) могут решить, какие свойства допустимы у потомства. Вправе ли общество влиять на решения социальной и личной евгеники, можно ли их регулировать? Есть ли рациональный способ проводить разницу между болезнью Тэя-Сакса, бета-талассемией, серповидной клеточной анемией (обе — заболевания крови) последствиями употребления матерью талидомида, болезнью Альцгеймера, фенилкетонурией, нетрадиционной сексуальной ориентацией (если когда-нибудь отыщется способ предсказывать ее), различными душевными заболеваниями, кистозным фиброзом, церебральным параличом, расщеплением позвоночника, ахондроплазией и связанными с ней нарушениями роста, гемофилией, синдромом Дауна, сердечно-сосудистыми заболеваниями, остеопорозом, патологическим ожирением и проч. В этой войне с нежелательными свойствами движение за права человека и равноправие окажется бессильным. Этому должен быть положен конец.
Хотя анонимный автор действительно ставит острые вопросы — например, относительно сексуальной ориентации, той или иной недоразвитости роста или избыточности веса, — защита некоторых из перечисленных заболеваний внушает тревогу, пусть она и продиктована законным и обоснованным страхом дискриминации лиц, страдающих этими недугами. Наш долг — создать гарантии, что такая «дискриминация» на самом деле будет нацелена на болезнь, а не на ее жертву.
Наследственные заболевания
Наследственные заболевания – это болезни, вызванные хромосомными и генными мутациями. Некоторые путают наследственные заболевания с врожденными. Действительно, врожденные болезни, то есть заболевания, с которыми рождается ребенок, могут быть наследственными, но их причиной может стать и какое-либо повреждающее внешнее воздействие на эмбрион или плод – инфекция, ионизирующее излучение, токсическое вещество. С другой стороны, не все наследственные заболевания врожденные, так как некоторые из них могут возникнуть и позднее, даже у взрослого человека. Появление наследственного заболевания не зависит от внешних причин и всегда обусловлено патологической мутацией.
Существуют еще и заболевания с наследственной предрасположенностью. Это сахарный диабет, атеросклероз, ожирение, язва желудка и двенадцатиперстной кишки и др. Они могут возникнуть у человека, родственники которого страдали этими патологиями, под влиянием внешних воздействий – неправильного питания, недостатка движения, сильного стресса (но это не означает, что такое обязательно случится).
Сегодня медицине известно около пяти тысяч наследственных заболеваний – генных и хромосомных.
Генные болезни
Большая часть всех наследственных заболеваний вызвана именно генными мутациями. К ним относятся ферментопатии – различные нарушения обмена веществ. Ферменты из-за мутации генов изменяют свои свойства или вообще не вырабатываются организмом, и в результате биохимическая реакция, в которой задействован данный фермент, не осуществляется.
К подобным наследственным заболеваниям относятся фенилкетонурия, гомоцистинурия, альбинизм, болезнь «кленового сиропа» (нарушен обмен аминокислот); галактоземия и фруктоземия (нарушения углеводного обмена); болезнь Тея–Сакса, плазматический липоидоз (нарушения жирового обмена); болезнь Коновалова–Вильсона (нарушения обмена металлов); болезнь Леша–Найхана (нарушения обмена пуринов).
Наследственные заболевания могут переходить из поколения в поколение, как, например, фенилкетонурия. При этой болезни организм не может усваивать фенилаланин, аминокислоту, ответственную за образование гормонов адреналина, тирозина, норадреналина. В результате возникают тяжелые поражения нервной системы, проявляющиеся нарушением двигательных функций, слабоумием.
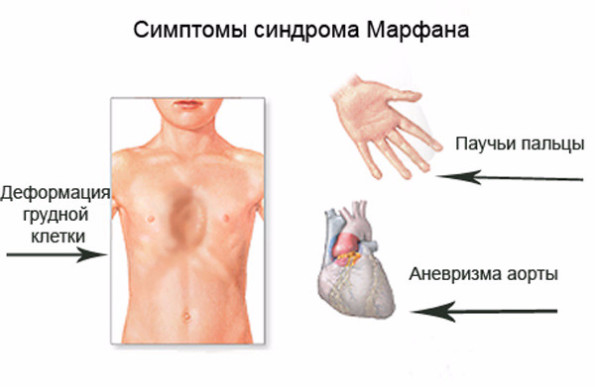
Синдром Марфана (арахнодактилия) – наследственное заболевание соединительной ткани вследствие мутации гена, отвечающего за синтез фибриллина. Болезнь затрагивает костно-мышечную систему, кожу, глаза, сердечно-сосудистую систему. Люди с синдромом Марфана отличаются худобой, высоким ростом, длинными руками и ногами («люди-пауки»), для них характерны сухость кожи, чрезмерная подвижность суставов, деформации позвоночника и грудной клетки. Они страдают пороками сердца, аневризмой аорты, подвывихом хрусталика. С интеллектом же у них все в порядке. Более того, синдромом Марфана страдали такие выдающиеся личности, как Авраам Линкольн, Никола Паганини, Шарль де Голль, Корней Чуковский.
Патологические мутации могут возникать и в процессе эмбрионального развития. Так, ахондроплазия – нарушение роста костей и карликовость – в 80% случаев вызвана новой мутацией, при этом в семье этой болезнью никто никогда не страдал.
Генной мутацией обусловлен и синдром Матрина–Белл (синдром ломкой X-хромосомы). Болезнь обнаруживается в детском возрасте и характеризуется умственной отсталостью.
Большинство наследственных заболеваний проявляется в детстве, но мутации генов могут дать знать о себе и в зрелом возрасте. Так, болезнь Альцгеймера, развивающаяся сравнительно рано, в 50 лет, обязана своим появлением именно генной мутации.

Хромосомные болезни
Эти болезни вызваны хромосомными и геномными мутациями, то есть изменением строения или числа хромосом. Они, как правило, появляются при формировании половых клеток. Часто такие мутации приводят к выкидышу или рождению мертвого младенца, в некоторых случаях ребенок рождается, но оказывается больным.
К хромосомным болезням относится всем известный синдром Дауна – в хромосомном наборе таких больных имеется лишняя хромосома. Для людей с синдромом Дауна характерны своеобразный внешний вид, умственная отсталость, пониженная сопротивляемость заболеваниям.
Синдром Шерешевского–Тернера – хромосомная болезнь, поражающая только женщин и заключающаяся в отсутствии одной половой хромосомы. У таких больных недоразвиты яичники, из-за чего внешние половые признаки сглажены: у них низкий рост, широкие плечи, короткие ноги, узкий таз. Характерная особенность – складки кожи, идущие от затылка на шею (шея сфинкса). Умственное развитие у таких больных остается в норме, но они отличаются эмоциональной неустойчивостью. У женщин с синдромом Шерешевского–Тернера отсутствуют менструации, и они не могут иметь детей.
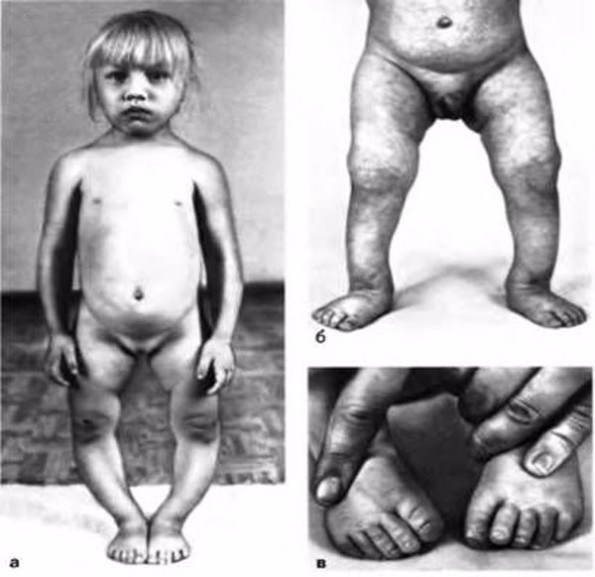
Синдром Клайнфельтера – мужская хромосомная аномалия. Она заключается в наличии у мужчины одной или нескольких женских половых хромосом, что и определяет «женственный» внешний вид больного – слабо развитую мускулатуру, узкие плечи, широкий таз. У мужчин с синдромом Клайнфельтера недоразвиты яички, и в результате сперматозоиды не образуются или образуются в очень малых количествах.
Часто мужчины узнают о своем заболевании, только когда решают завести детей. После исследований выясняется, что бесплодие вызвано именно синдромом Клайнфельтера.
Синдром кошачьего крика, или синдром Лежена, вызван нарушением в структуре 5-й хромосомы. Назван синдром из-за необычного плача детей, высокого, пронзительного, напоминающего мяуканье кошки, что связано с дефектом в развитии гортани. Дети с этим синдромом рождаются с микроцефалией (маленькая голова), они страдают умственной неполноценностью, у них обнаруживается большое число отклонений в развитии различных органов, тяжелые осложнения. Большинство из них умирают в раннем возрасте.
Профилактика наследственных заболеваний
Сегодня часть наследственных заболеваний можно обнаружить еще до рождения ребенка с помощью молекулярно-генетических исследований. Конечно, некоторые такие исследования небезопасны, поэтому их выполняют только при необходимости, когда женщина находится в группе риска: в семье имеются случаи наследственных заболеваний, первый ребенок родился больным, если женщина рожает после 35 лет (риск рождения ребенка с синдромом Дауна) и др. Но лучше, если оба родителя, пройдут генетическое исследование еще на стадии планирования беременности и определят, насколько велик у них риск рождения ребенка с наследственным заболеванием.
- http://www.vechnayamolodost.ru/articles/biomedicin/molekulyarnie_bombi_s_programmnim_upravleniem/
- http://sovets.net/12417-nasledstvennye-zabolevaniya.html
- http://www.neboleem.net/stati-o-zdorove/7816-nasledstvennye-zabolevanija.php
- http://bio.wikireading.ru/935
- http://www.medsovet.info/articles/3291